Aging bodies become increasingly burdened over time with dysfunctional cells resistant to apoptotic or other clearance. The most well-known of these are so-called “senescent” cells, originally characterized by Leonard Hayflick as mitotic cells that reached growth arrest after a limited replicative lifespan (later associated with telomere attrition) under unphysiological conditions in culture. Later research has revealed that few cells reach a “senescent” state through sheer replicative exhaustion: instead, senescence has emerged as a programmed response to DNA damage or oncogenic stress, and as part of the resolution of wound healing.(1) Unfortunately, the near-term benefits of these functions — in preventing damaged cells from progressing to cancer, and in preventing fibrosis –are coupled to deleterious long-term consequences, whose effects worsen as the burden of such cells rises with aging. First, the loss of mitotic competence of stem cells denies proliferative tissues of the capacity for renewal. Secondly, the secretory and other phenotypes of such cells progressively derange local and systemic metabolism and tissue function, rendering tissues more vulnerable to metastasis, promoting systemic inflammation, and otherwise impairing tissue function.(1-4)
To bypass the disruptive effects of the age-related accumulation of senescent cells, some investigators are working on possible ways to manipulate the signaling pathways involved in enforcing the senescent phenotype. This approach bears with it great risks, however, because of the very purposes of senescence to which allusion was made above: returning senescent cells to their normal differentiated function and replicative capacity could lead cells bearing oncogenic mutations to progress into metastatic disease, and aberrant resumption of the wound-healing response leading to fibrosis.(1,4) The regenerative engineering solution to this dilemma is therefore the ablation of such cells, to eliminate their contribution to age-related loss of homeostasis without reactivating the more acute risks against which the senescence machinery was activated in the first place.(5)
As widely covered in the mainstream press, a successful proof-of-principle study for this rejuvenation biotechnology has now been performed.(6)
The study was performed using several founder lines, bred onto a background strain of mice hypomorphic for BubR1 (BubR1H/H), a key component of the mitotic checkpoint machinery. Principal investigator Jan van Deursen, Professor of Biochem/Molecular Biology and of Pediatrics at the Mayo Clinic location in Minnesota, had already discovered(7) that BubR1H/H mice “have a markedly shortened lifespan and exhibit a variety of age-related phenotypes, including infertility, lordokyphosis, sarcopenia, cataracts, [subcutaneous] fat loss, cardiac arrhythmias, arterial wall stiffening, impaired wound healing and dermal thinning.”(6) Some, but not all, of these phenotypes were associated with a high age-related incidence in senescent (p16Ink4a-positive) cells.(6-8) and van Deursen and colleagues had already demonstrated that breeding BubR1H/H mice onto a p16Ink4a homozygous-null genetic background attenuated their development of p16Ink4a-senescent cell-associated aging phenotypes and modestly increased their very low survivorship.(8) Imputation of these results specifically to the animals’ age-related, low-BubR1-driven rise in p16Ink4a-expressing senescent cells was, however, limited: limited by the very nature of so-called “accelerated aging” models such as BubR1H/H,(9) and limited by the lifelong, global absence of p16Ink4a expression in the backcrossed mice.
Seeds of Destruction and Renewal
To impute aging phenotypes directly to p16Ink4a-expressing senescent cells, van Deursen and colleagues with expertise in the aging and senescence of the relevant tissues developed and tested the effects of a pharmacologically-inducible system for the ablation of p16Ink4a-expressing cells. To create this system, investigators modified an approach used in earlier research, in which mice were bred with a variant on the Gene-Directed Enzyme Prodrug Therapy (or “suicide gene”) paradigm,(11) using a drug (AP20187) that activated fusion protein apoptosis machinery in cells in which the macrophage- and adipocyte-specific minimal Fabp4 promoter was transcriptionally active.(10) To generate mice in which p16Ink4a-expressing cells could be similarly selectively ablated, van Deursen’s team substituted a fragment of the p16Ink4a gene promoter for the Fabp4 promoter, thereby generating BubR1H/H;INK-ATTAC mice.(6) In such mice, then, p16Ink4a would still be under normal physiological regulation, and still be induced in an abnormally high number of cells due to the mitotic checkpoint dysfunction caused by BubR1 hypomorphism, leading to the same abnormally-rapid accumulation of high burdens of p16Ink4a-expressing senescent cells — but administration of AP20187 would induce apoptosis selectively in such cells, purging the animals’ tissues of senescent cells while leaving non-senescent cells unscathed.
Testing the System
A range of in vitro and in vivo tests was used to rigorously confirm the selectivity and sensitivity of the system’s activation in, and ablation of, p16Ink4a-positive senescent cells.(6) In early-aging (2-mo old) BubR1H/H;INK-ATTAC mice, but not young (3-wk-old) mice, transcripts of the system and of reporter green fluorescent protein “were significantly elevated in [subcutaneous] adipose tissue, skeletal muscle and eye, but not in tissues in which endogenous p16Ink4a is not induced, including liver and heart.”(6) Moreover, subcutaneous adipose of prematurely-aged transgenic mice exhibited high levels of staining for the senescence marker senescence-associated-β-galactosidase (SAβ-gal) and expressed high levels of several established markers of senescence, including p21, p19, interleukin-6, (insulin-like growth factor binding protein-2 (Igfbp2), and Pai-1; primary BubR1H/H;INK-ATTAC mouse embryonic fibroblasts forced artificially into senescence by oncogenic Ras or serial passage exhibited a subpopulation that was both GFP+ and stained positively SAβ-gal. BubR1H/H;INK-ATTAC muscle cells and lens did not stain for SAβ-gal, but did exhibit selective induction of the “senescence genes.”(6)
When BubR1H/H;INK-ATTAC mouse bone marrow cells were pushed into senescence in vitro by the PPAR-γ-activating drug rosiglitazone, a subpopulation of the cells exhibited high levels of INK-ATTAC expression and GFP, coupled with SAβ-gal staining; subsequent to treatment with the INK-ATTAC activating drug, these cells rapidly entered into apoptosis, and within 48 h were either destroyed or in the cell death process.(6)
Ablation of Senescent Cells Retards Age-Related Tissue Degeneration
As a first approach, the investigators abrogated the premature age-related rise of p16Ink4a-senescent cell burden in the tissues of BubR1H/H;INK-ATTAC mice by initiating a lifelong course of senescent-cell-ablating AP20187 treatment at weaning. At 9-10 mo of age, such mice were then compared a to age-matched cohorts of untreated BubR1H/H;INK-ATTAC mice, and to BubR1H/H mice lacking the INK-ATTAC system for selective ablation of p16Ink4a-expressing cells. Relative to both control cohorts of the same age, 9-10 mo old treated mice exhibited dramatically more youthful tissues. Consistent with earlier results, their burden of p16Ink4a-positive senescent cells in muscle, eye, and adipose tissues were far lower. Their muscle fibers had larger diameters, their treadmill endurance was greater and they covered more distance on them. Treated mice had fewer cataracts, and less lordykyphosis. And they suffered less lipoatrophy, with larger fat deposits in multiple depots, higher individual adipocyte volumes, and more proliferating cells marked with BrDU (see Figure 1, below).(6) No treatment-related adverse events presented themselves.(6)

Figure 1: Amelioration of “Premature Aging” Phenotypes in Treated and Untreated BubR1H/H;INK-ATTAC Mice. AP=AP20187 treatment. Reproduced from (6). © Nature Publication Group.
Importantly, “premature aging” phenotypes observed in BubR1H/H mice over time, but that are in tissues where p16Ink4a-positive senescent cells do not accumulate with aging, were not alleviated by drug treatment. Thus, these animals exhibit premature cardiac arrhythmias and stiffening of the arterial wall, and cardiac failure appears to be the main cause of death; yet these tissues are not burdened with an abnormally-high burden of p16Ink4a-senescent cells, and accordingly, ablation p16Ink4a-positive senescent cells in these animals had little tissue-specific or survivorship phenotypic impact.(6)
Following this initial test of abrogating the early, age-related rise in p16Ink4a-expressing cell burden, the investigators probed the effects of leaving BubR1H/H;INK-ATTAC to undergo 5 months of rapid “premature aging” (and thus, to the attendant accumulation of high levels of p16Ink4a-positive cells and onset of “early-aging” phenotypes), and only then inducing ablation of senescent cells with the INK-ATTAC drug-activated system (see Figure 2 (g) below).(6) At that point, the animals’ cataracts had already reached peak age-related severity, and remained stable after 5 months of further aging irrespective of treatment. But muscle fibers that continued to atrophy over the ensuing 5 months in control animals remained at their more youthful diameters in animals whose p16Ink4a-positive senescent cells had been ablated, and treadmill times, distance traveled, and work outputs were maintained at substantially more youthful levels (Figure 2, (a) and (b) below). Similarly, the degeneration of adipose tissue cells and depots that occurred over the course of the next 5 months in control animals was virtually abrogated, leaving 10 mo-old animals with substantially the same subcutaneous and other fat tissue (in depth and in cell volume) as they had enjoyed in their relative youth, when treatment was first initiated.(6)
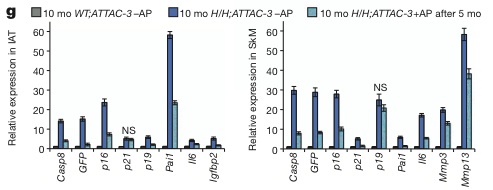
Figure 2: Ablation of Senescent p16Ink4a-Expressing Cells Maintains Youthful Muscle and Adipose Tissues. Reproduced from (6). © Nature Publication Group.
Rejuvenation Implications
As noted above, studies involving the use of putative “premature aging” models must be interpreted with caution, as the designation inevitably involves an element of petitio principii: from a subset of similar phenotypes are drawn conclusions of similar aetiology, and from this, further conclusions about the “normal” degenerative aging process (and its biomedical amelioration) are too-readily drawn before the thesis itself has first been established.(9) Indeed, the degenerative aging process is by definition one in which the organism progressively accumulates damage to its cellular and molecular components over time, so any genetic or environmental factor that leads to a greater burden of such damage will bear some resemblance to the aging phenotype, irrespective of the causal origin of the defect or its relationship to “normal” aging.
In the case of this new report,(6) however, while caution is still merited, the nature of the intervention used makes the study relatively free of such complications. The investigators did not simply modulate or normalize the very thing that the mutation (in this case, to the mitotic checkpoint component BubR1) itself disrupts, as in other widely-publicized studies involving putative “accelerated aging” (eg. (12,13)). Rather, the defective checkpoint system was left to proceed, and one of its downstream consequences, which was still under normal regulation — and one known to be directly induced by the normal degenerative aging process — was reversed at the structural level, by clearing out the p16Ink4a-positive senescent cells that had accumulated to an abnormal degree in their tissues. This left some aspects of the abnormal “progeroid” phenotype in these organisms (the cardiovascular defects) intact, but illustrated the dysfunctional consequences of having tissues riddles with such cells. While still of abnormal origin, there is no strong reason to think that the ongoing effects of a rising burden of such cells would not be similar — and thus, that the effects of ablating such cells are uninformative about the effects of a similar intervention in “normally” aging bodies.*
The links to aging phenotypes, and their near-arrest by ablation of p16Ink4a-expressing senescent cells, appear to be dramatic illustrations of the deleterious effects of the age-related rise in the burden of senescent cells in genetically-intact mammals. The fact that it was the removal of such cells from aging tissues that arrested multiple aging phenotypes is of special importance to the rejuvenation biotechnology approach to preventing and reversing age-related disease and disability: it clearly identifies the damage itself, rather than the abnormal function of either p16Ink4a (which was under normal, physiological regulation, rather than being pharmacologically modulated, or knocked out as in their previous report(8)) or BubR1 (mutation of which, and its direct metabolic sequelae, was not affected by the intervention).
And there are reasons to believe that the resulting arrest of multiple aspects of tissue aging by removal of p16Ink4a-expressing senescent cells would indeed translate into the tissues of genetically-intact mice — or humans.
Sarcopenia
The fact — and disabling and fatal consequences — of age-related decline in muscle quality and quantity is widely known, but the contribution of “senescent” cells to this degenerative process is not. While some studies have reported no decline in satellite cells (muscle progenitor cells) with aging, others (eg. (14,15)) have found age-related satellite cell attrition consistent with the senescence of a subset thereof; moreover, one such study (15) reported that decreases in the number and quality of satellite cells with aging are reliably associated with elevated expression of p16Ink4a (contrary to (14)), and with secretory and proteomic abnormalities consistent with a rising burden of senescent cells. Consistent with a causal relationship, (16) reports that the prevalence of limited physical functioning in aging varies depending on p16Ink4a allelic variation, consistent with variations in rate of stem cell attrition with senescence.
There is therefore good reason to expect that the profound arrest of sarcopenic phenotypes observed in p16Ink4a-senescent cell ablated BubR1H/H “premature aging” mice would translate into the human case.
Lipoatrophy
While less well-known (masked as it is and placed out of focus by the overall age-related body composition shift from lean mass to adiposity), there is none the less significant age-related subcutaneous lipoatrophy in aging, most visibly in the sunken appearance of the face. Part of this is a pathological redistribution of adipose from the subcutaneous to the visceral depot, but it now emerges that the subcutaneous depot becomes qualitative as well as quantitatively abnormal in the degenerative aging process also suffers genuine age-related lipoatrophy and lipodystrophy — and that p16Ink4a-driven cellular senescence is at the heart of it.
Subcutaneous adipose tissue contributes to maintenance of insulin sensitivity and other aspects of metabolic homeostasis, through the production of adipose-specific endocrine factors such as adiponectin. Surgical removal of subcutaneous fat reduces adiponectin levels and insulin sensitivity, and transplantation of subcutaneous fat increases both.(17) Slow-aging growth hormone receptor knockout (GHRKO) mice are obese, but highly insulin sensitive: in such animals, surgical removal of visceral adipose tissue impairs insulin secretion and peripheral insulin action, in part by reducing adiponectin production. (21) Moreover, while the link between excessive visceral adipose tissue and age-independent diabetes and metabolic syndrome widely known, recent studies suggest instead that it is the accumulation of senescent subcutaneous adipocyte progenitors — and their abnormal metabolic function — that drives similar diabetes-like phenotypes during the “normal” aging process.(3,18; cf. 19,20) Even in visceral fat, it has recently emerged that the obesity-driven rise in inflammation and insulin resistance is associated with an abnormal accumulation of senescent cells, albeit senescent endothelial cells rather than adipocytes.(20) It was this emerging line of research that van Deursen’s collaborator Dr. James Kirkland presented at the fifth annual Strategies for Engineered Negligible Senescence Conference (SENS5) in September of this year,(18) and it was his expertise in the senescence of adipose tissue that he contributed to the new report on the effects of ablating such cells.(6)
Again, then, there is significant evidence consistent with a role of cellular senescence in age-related lipodystrophy and lipoatrophy, and for the benefits observed in treated mice in these studies to translate into aging humans. It is unfortunate that the investigators did not assess insulin secretion, insulin action, or systemic inflammation in early-aging BubR1H/H;INK-ATTAC mice, with and without ablation of senescent adipose cells, but reasonable to be optimistic that doing so would yield some normalization of age-related metabolic abnormalities.
Cataract
There is only the most tentative of evidence suggesting a link between cellular senescence and cataract in “normal” aging.(22) Absence of evidence is, however, not evidence of absence, and certainly the inflammatory secretory profile of senescent cells would, if present, likely accelerate the degenerative course of the disease.
Cancer
A great deal of evidence has now been amassed that stromal cell senescence plays an important role in laying the groundwork for tumor metastasis, promoting cell proliferation with inflammatory cytokines, encouraging angiogenesis, and degrading the tumor-suppressive action of an intact extracellular matrix.(1) One clear disadvantage of using these “early-aging” mice is that they die too early to develop cancer — too early for ablation of p16Ink4a-positive senescent cells to impact the course of the disease. It would indeed be of great interest to see whether ablation of stromal p16Ink4a-expressing senescent cells, in otherwise genetically intact INK-ATTAC animals without existing tumors, would lower the animals’ risk for cancer, and put any tumors they might develop on a less malevolent trajectory, than untreated mice. It should be noted, however, that while a study on senescent cell ablation in genetically normal mice would provide at least some evidence on the effect of senescent cells (and their ablation) on promoting cancer, even such a study would likely show less effect than could be anticipated in a large mammal model, since even normally-aging mice rarely suffer metastatic disease to the extent of aging humans, as sheer primary tumor volume is generally sufficient to be fatal to mice.
Other Tissues
As the investigators note, the rapid age-related arterial stiffening and cardiac arrhythmias that appear to be at cause for the majority of deaths in BubR1H/H mice were not attenuated by ablating p16Ink4a-expressing senescent cells — but these tissues had little burden of such cells, so this finding reinforces the conclusion that the multiple aging phenotypes arrested in these mice when senescent cells were ablated is attributable specifically to the removal of their baleful influence on local tissues. On the other hand, there are many other tissues — notably, the kidney and articular cartilage — where p16Ink4a-expressing senescent cells appear to be a contributing factor to human and murine degenerative aging, but which were not evaluated in treated or control mice in this study, and it would be of interest to see the effects of ablation of p16Ink4a-positive senescent cells.
Moreover, there are yet other cell types — such as visceral adipose tissue macrophages and cytotoxic CD8+ T-cells — in which the age-related supernumerary accumulation of dysfunctional and apoptosis-resistant cells appears to play a highly deleterious role on tissue function, but where the cells are not “senescent” cells in the classical sense of p16Ink4a expression and the senescence-associated secretory profile observed in senescent fibroblasts. This study (6) cannot provide evidence directly on the effects of ablating such cells, but it does provide an analogous proof-of-concept for the approach. SENS Foundation is funding ongoing work in the lab of Dr. Janko Nikolich-Zugich to investigate the effects of clearance of anergic, “senescent” cytotoxic CD8+ T-cells on immunosenescence,(22) and is interested in the targeting of other such cells.(2)
Arrest vs Reversal
In the new study, p16Ink4a-expressing senescent cells were ablated either at weaning or some months later, and assessed several months after the initial intervention. Remarkably enough, the removal of such cells arrested tissue degeneration, holding the muscles and adipose tissue (and, when administered before cataract was mature, lens opacification) at approximately the same relatively youthful condition prevalent when the inducing drug was first administered (see eg. Fig 2(a) above).(6) This is consistent with the deleterious effect of such cells on tissue function, and with the researchers’ conclusion that “the observed improvements in skeletal muscle and fat of late-life treated 10-month-old BubR1H/H;INK-ATTAC-5 mice reflect attenuated progression of age-related declines rather than a reversal of ageing”.(6) However, it would be useful to see a more thorough analysis of the effect of ablating p16Ink4a-expressing senescent cells, and whether there may instead be evidence of a short-term rejuvenation of tissue function that is slowly lost over time to rising levels of other kinds of aging damage that INK-ATTAC activation does not address. Indeed, as illustrated by the lack of effect of p16Ink4a-expressing cell ablation on lifespan, and by the ongoing degeneration of tissues (such as the heart) in which p16Ink4a-postive senescent cells are not a driver of “early aging,” true rejuvenation requires a comprehensive suite of rejuvenation biotechnologies to remove all forms of aging damage from the aging body.
Translation for Human Rejuvenation Biotechnologies
The investigators boldly, but rightly, conclude that
Our proof-of-principle experiments demonstrate that therapeutic interventions to clear senescent cells or block their effects may represent an avenue for treating or delaying age-related diseases and improving healthy human lifespan.(6)
How might the results of this intervention be translated for human rejuvenation therapies?
There is already evidence that senescent cells are targeted by the innate immune system.(24-28) Dr. Judith Campisi, in fact, has found that activating NKG2D receptors on natural killer (NK) cells engage MHC class I chain-related protein A and B (MICA/B) ligands on senescent cells, leading to their NK-induced apopotsis and subsequent clearance.(29) MICA/B ligands are also used to activate tumor cell destruction by NK cells via NKG2G binding, and tumors evolve resistance by several mechanisms to reduce cell-surface MICA abundance;(30) however, the natural selection mechanisms that drive the evolution of such defenses do not apply to growth-arrested cells. Dr. Campisi has found instead that a minority of senescent cells evade destruction by secreting high levels of matrix metalloproteinases (MMPs), which cleave MICA/B ligands and thereby prevent NKG2D binding.(29) This has led to the hypothesis that the great majority of such cells are destroyed over the lifetime by innate immunity, and that the specific senescent cells that do accumulate with aging are precisely those who had variants that allow MMP overexpression, in a kind of “one-off,” very temporally-extended kind of selection process. Potentially, a kind of intervention that could overcome this resistance to endogenous clearance mechanisms would allow for the purgation of senescent cells from aging tissues.
SENS Foundation is currently funding work by PhD Candidate Kevin Perrott in Campisi’s laboratory, screening compounds for their effectiveness in mitigating the negative impact of cells exhibiting the classical senescence-associated secretory phenotype (SASP)(1) following senescence induced by treatment with 10 gray of ionizing radiation by selective induction of apoptosis or modulating their secretions. To date, a screen of the collection of FDA-approved drugs in the Prestwick Library has identified a handful of potential candidates which have demonstrated effectiveness at lowering secretion of IL-6, a component of SASP whose concentration tends to rise systemically with aging and here used as a preliminary marker of SASP as a phenotype. In particular, Perrott has recently identified some members of a class of compounds that lower the SASP in irradiated-senescent cells, without reversing growth arrest, and he is currently investigating the mechanisms of this phenomenon.
It is clear that there is substantial distance yet to be traveled. Multiple cell types acquire distinctive “senescent” phenotypes on a cell-type-specific basis, and will require ablation to achieve comprehensive rejuvenation. However, this important proof-of-principle from Dr. van Deursen’s laboratory, and the key validation of the scope of the effects of ablation of these particular senescent cells facilitated by his collaboration with the LeBrasseur and Kirkland labs at Mayo, stands as a key landmark in moving toward the removal of their baleful influence on aging tissues. As ever, SENS Foundation is committed to making investments in critical-path research to advance this key but heretofore-neglected line of biomedical research out of the laboratory, into the clinic, and to uniting the multiple strands of rejuvenation biotechnologies into a comprehensive panel for the restoration of the health, vigor, and open futures of aging humanity.
* The principle caveat would be that the interaction of senescence with defective mitotic checkpoint function within such cells, and the effects upon their neighbors of their state and of their senescence-associated secretory phenotype, would very likely cause some phenomena that would not be observed in p16Ink4a–senescent cells or in their effects on neighbors.
References
1: Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. 2011 Feb 21;192(4):547-56. Epub 2011 Feb 14. Review. PubMed PMID: 21321098; PubMed Central PMCID: PMC3044123.
2: Burton DG. Cellular senescence, ageing and disease. Age (Dordr). 2009 Mar;31(1):1-9. Epub 2008 Sep 4. PubMed PMID: 19234764; PubMed Central PMCID: PMC2645988.
3: Tchkonia T, Morbeck DE, Von Zglinicki T, Van Deursen J, Lustgarten J, Scrable H, Khosla S, Jensen MD, Kirkland JL. Fat tissue, aging, and cellular senescence. Aging Cell. 2010 Oct;9(5):S26(Abs 49). doi: 10.1111/j.1474-9726.2010.00608.x. Epub 2010 Aug 15. Review. PubMed PMID: 20701600; PubMed Central PMCID: PMC2941545.
4: Naesens M. Replicative senescence in kidney aging, renal disease, and renal transplantation. Discov Med. 2011 Jan;11(56):65-75. Review. PubMed PMID: 21276412.
5: de Grey AD. Foreseeable pharmaceutical repair of age-related extracellular damage. Curr Drug Targets. 2006 Nov;7(11):1469-77. Review. PMID: 17100587 [PubMed – indexed for MEDLINE]
6: Baker DJ, Wijshake T, Tchkonia T, Lebrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM. Clearance of p16(Ink4a)-positive senescent cells delays ageing-associated disorders. Nature. 2011 Nov 2. doi: 10.1038/nature10600. [Epub ahead of print] PubMed PMID: 22048312.
7: Baker DJ, Jeganathan KB, Cameron JD, Thompson M, Juneja S, Kopecka A, Kumar R, Jenkins RB, de Groen PC, Roche P, van Deursen JM. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat Genet. 2004 Jul;36(7):744-9. Epub 2004 Jun 20. PubMed PMID: 15208629.
8: Baker DJ, Perez-Terzic C, Jin F, Pitel K, Niederländer NJ, Jeganathan K, Yamada S, Reyes S, Rowe L, Hiddinga HJ, Eberhardt NL, Terzic A, van Deursen JM. Opposing roles for p16Ink4a and p19Arf in senescence and ageing caused by BubR1 insufficiency. Nat Cell Biol. 2008 Jul;10(7):825-36. Epub 2008 May 30. PubMed PMID: 18516091; PubMed Central PMCID: PMC2594014.
9: Miller RA. ‘Accelerated aging’: a primrose path to insight? Aging Cell. 2004 Apr;3(2):47-51. Review. PubMed PMID: 15038817.
10: Pajvani UB, Trujillo ME, Combs TP, Iyengar P, Jelicks L, Roth KA, Kitsis RN, Scherer PE. Fat apoptosis through targeted activation of caspase 8: a new mouse model of inducible and reversible lipoatrophy. Nat Med. 2005 Jul;11(7):797-803. Epub 2005 Jun 19. PubMed PMID: 15965483.
11: Both GW. Gene-directed enzyme prodrug therapy for cancer: a glimpse into the future? Discov Med. 2009 Oct;8(42):97-103. Review. PubMed PMID: 19833053.
12: Sahin E, Colla S, Liesa M, Moslehi J, Müller FL, Guo M, Cooper M, Kotton D, Fabian AJ, Walkey C, Maser RS, Tonon G, Foerster F, Xiong R, Wang YA, Shukla SA, Jaskelioff M, Martin ES, Heffernan TP, Protopopov A, Ivanova E, Mahoney JE, Kost-Alimova M, Perry SR, Bronson R, Liao R, Mulligan R, Shirihai OS, Chin L, DePinho RA. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature. 2011 Feb 17;470(7334):359-65. Epub 2011 Feb 9. Erratum in: Nature. 2011 Jul 14;475(7355):254. PubMed PMID: 21307849.
13: Jaskelioff M, Muller FL, Paik JH, Thomas E, Jiang S, Adams AC, Sahin E, Kost-Alimova M, Protopopov A, Cadiñanos J, Horner JW, Maratos-Flier E, Depinho RA. Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice. Nature. 2011 Jan 6;469(7328):102-6. Epub 2010 Nov 28. PubMed PMID: 21113150; PubMed Central PMCID: PMC3057569.
14: Carlson ME, Suetta C, Conboy MJ, Aagaard P, Mackey A, Kjaer M, Conboy I. Molecular aging and rejuvenation of human muscle stem cells. EMBO Mol Med. 2009 Nov;1(8-9):381-91. PubMed PMID: 20049743; PubMed Central PMCID: PMC2875071.
15: G. Butler-Browne, M.-C. LeBihan, A. Bigot, D. Furling, F. Svinartchouk, D. Bechet, V. Mouly. Identification of biomarkers of human muscle aging and senescence. Rejuvenation Res. 2007 Sep;10(Suppl1):S22(Abs 14).
16: Melzer D, Frayling TM, Murray A, Hurst AJ, Harries LW, Song H, Khaw K, Luben R, Surtees PG, Bandinelli SS, Corsi AM, Ferrucci L, Guralnik JM, Wallace RB, Hattersley AT, Pharoah PD. A common variant of the p16(INK4a) genetic region is associated with physical function in older people. Mech Ageing Dev. 2007 Mar 27; [Epub ahead of print] PMID: 17459456 [PubMed – as supplied by publisher]
17: Tran TT, Yamamoto Y, Gesta S, Kahn CR. Beneficial effects of subcutaneous fat transplantation on metabolism. Cell Metab. 2008 May;7(5):410-20. PubMed PMID: 18460332; PubMed Central PMCID: PMC3204870.
18: Kirkland JL. Aging, Adipose Tissue, and Cellular Senescence. Abstracts of Strategies for Engineered Negligible Senescence (SENS) Fifth Conference. August 31-September 4, 2011. Cambridge, United Kingdom. Rejuvenation Res. 2011 Aug;14 Suppl 1:S11-45. PubMed PMID: 21847798.
19: Minamino T, Orimo M, Shimizu I, Kunieda T, Yokoyama M, Ito T, Nojima A, Nabetani A, Oike Y, Matsubara H, Ishikawa F, Komuro I. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat Med. 2009 Sep;15(9):1082-7. Epub 2009 Aug 30. PubMed PMID: 19718037.
20: Villaret A, Galitzky J, Decaunes P, Estève D, Marques MA, Sengenès C, Chiotasso P, Tchkonia T, Lafontan M, Kirkland JL, Bouloumié A. Adipose tissue endothelial cells from obese human subjects: differences among depots in angiogenic, metabolic, and inflammatory gene expression and cellular senescence. Diabetes. 2010 Nov;59(11):2755-63. Epub 2010 Aug 16. PubMed PMID: 20713685; PubMed Central PMCID: PMC2963533.
21: Bartke A. Effects of Calorie restriction in long-lived mice. Presentation at CR VII, Seventh Calorie Restriction Society Conference, Las Vegas, NV, October 26-29 2011.
22: Zhang ZF, Zhang J, Hui YN, Zheng MH, Liu XP, Kador PF, Wang YS, Yao LB, Zhou J.Up-Regulation of NDRG2 in Senescent Lens Epithelial Cells Contributes to Age-Related Cataract in Human. 2011;6(10):e26102. Epub 2011 Oct 17. PubMed PMID: 22043305; PubMed Central PMCID: PMC3197158. doi:10.1371/journal.pone.0026102
23: Smithey MJ, Renkema KR, Rudd BD, Nikolich-Žugich J. Increased apoptosis, curtailed expansion and incomplete differentiation of CD8+ T cells combine to decrease clearance of L. monocytogenes in old mice. Eur J Immunol. 2011 May;41(5):1352-64. doi: 10.1002/eji.201041141. Epub 2011 Apr 14. PubMed PMID: 21469120.
24: Chien Y, Scuoppo C, Wang X, Fang X, Balgley B, Bolden JE, Premsrirut P, Luo W, Chicas A, Lee CS, Kogan SC, Lowe SW. Control of the senescence-associated secretory phenotype by NF-{kappa}B promotes senescence and enhances chemosensitivity. Genes Dev. 2011 Oct 15;25(20):2125-36. Epub 2011 Oct 6. PubMed PMID: 21979375.v
25: Radaeva S, Sun R, Jaruga B, Nguyen VT, Tian Z, Gao B. Natural killer cells ameliorate liver fibrosis by killing activated stellate cells in NKG2D-dependent and tumor necrosis factor-related apoptosis-inducing ligand-dependent manners. Gastroenterology. 2006 Feb;130(2):435-52. PubMed PMID: 16472598.
26 : Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, Yee H, Zender L, Lowe SW. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008 Aug 22;134(4):657-67. PubMed PMID: 18724938; PubMed Central PMCID: PMC3073300.
27: Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007 Feb 8;445(7128):656-60. Epub 2007 Jan 24. Erratum in: Nature. 2011 May 26;473(7348):544. PubMed PMID: 17251933.
28: Soriani A, Zingoni A, Cerboni C, Iannitto ML, Ricciardi MR, Di Gialleonardo V, Cippitelli M, Fionda C, Petrucci MT, Guarini A, Foà R, Santoni A. ATM-ATR-dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK-cell susceptibility and is associated with a senescent phenotype. Blood. 2009 Apr 9;113(15):3503-11. Epub 2008 Dec 19. PubMed PMID: 19098271.
29: Campisi J. New tricks for old cells. Understanding Aging: Biomedical and Bioengineering Approaches. 2008 Jun 27-26, UCLA.Program p.4-5.
30: Fuertes MB, Girart MV, Molinero LL, Domaica CI, Rossi LE, Barrio MM, Mordoh J, Rabinovich GA, Zwirner NW. Intracellular retention of the NKG2D ligand MHC class I chain-related gene A in human melanomas confers immune privilege and prevents NK cell-mediated cytotoxicity. J Immunol. 2008 Apr 1;180(7):4606-14. PubMed PMID: 18354183