ApoptoSENS
Removing Dysfunctional Cells
At conception, all of our cells have wide-open potential to develop into many different kinds of cell, including liver, immune, muscle, or brain neuronal cells. But even after progressing into their respective specific types, cells continue to adapt to changing internal and external conditions. Some of these adaptations sustain the function of the tissue and the life of the organism in the short term, but come back to bite us when they persist with age. As the number of these cells in our tissues rises with age, they begin to reach levels that impair tissue function and are harmful to health. Examples include:
Classic Senescent Cells
The original and most well-studied class of aberrant cells that accumulate in aging tissues are “senescent” cells. The senescence program is activated in cells that undergo risky changes in their DNA expression that put them on a path toward becoming cancerous. It is also activated in cells involved in the late stages of wound response, and in specific aspects of fetal development. In these contexts, the senescence machinery shuts down cellular reproduction and send out signals that remodel local connective tissue and attract cells from the immune system to eliminate them from the tissue.

Unfortunately, this immune clearance process is never totally exhaustive, and it may also become less effective as we age. The number of such “leftover” senescent cells in youthful tissues is so small as to be harmless, but after decades of accumulation, there are enough of them in the aging tissue that their abnormal metabolic state begins to pose a threat to surrounding, healthy cells. The rising burden of senescent cells in aging tissues makes them more vulnerable to the spread of cancer, contributes to inflammation, and is linked to multiple diseases of aging.
The strongest evidence that senescent cells are pathological is what happens when these “zombie” cells are purged out of aging tissues by proof-of-concept rejuvenation biotechnologies. Destroying senescent cells dramatically rejuvenates aging animals, and prevents, ameliorates, or reverses animal models of age-related disease (see more under “The Rejuvenation Biotechnology Solution,” below).
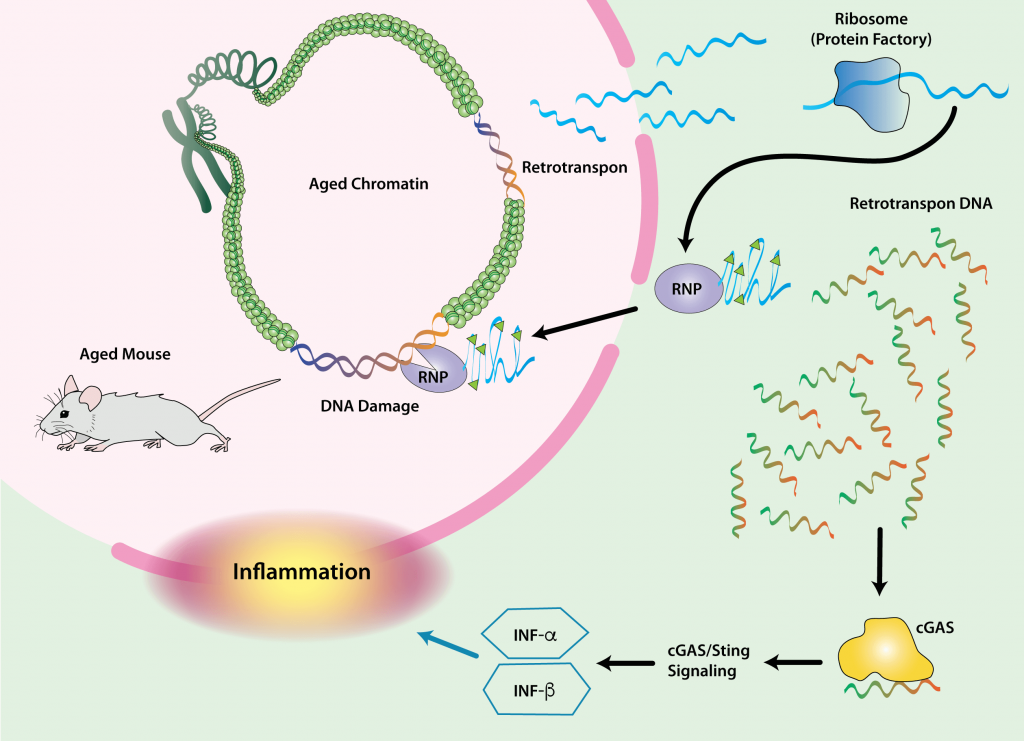
Cells with Reactivated Retrotransposons
Cells also become dysfunctional with age due to the reactivation of the suppressed remains of ancient infections lurking in our genomes. More than half of the human genome is comprised of invasive genetic material left behind by viruses, and an important subset of this material encodes retrotransposons: genetic machinery that can still be reactivated, replicate, and spread itself through the genome.
These reactivation events can have serious consequences for the host cell and the aging body. They can cause mutations in our functional genes, or disrupt the expression of non-mutated genes, leading to cancer, cellular self-destruction (apoptosis), and cellular senescence. In fact, there is a sinister relationship between retrotransposon reactivation and cellular senescence. The late stages of a cell’s transition into senescence can itself unleash retrotransposons — and when it happens, the reactivation of these retroviruses enforces the production of SASP.
Because of the ever-present threat of retrotransposon reactivation, the cell has machinery to keep them repressed. The importance of this machinery as a defense against degenerative aging is supported by the fact that multiple interventions known to slow aging in mice further tighten the screws on retrotransposons — as apparently does long-term exercise in humans. But this machinery falters with age and is temporarily suspended during periods when the cell is busy responding to genetic damage.
When all else fails, the eruption of reactivated viral genetic material in cells with reactivated retrotransposons triggers the production of the inflammatory signaling molecule interferon gamma, through which affected cells can either commit “cellular suicide” (apoptosis) or “turn themselves in” for destruction by the immune system. However, neither of these clearance mechanisms is foolproof, leading to accumulation of damaged interferon-signaling cells with age — another way that retrotransposon-activated cells can serve as a source of chronic inflammation, beyond their role in the SASP. Accordingly, reactivation of one type of retrotransposon has been tied to tissue inflammation in aging, and suppressing retrotransposon activity with drugs has been shown to inhibit tissue inflammation in aging mice.
Dysfunctional Cells in Fat Tissue
Even in lean, physically active, and otherwise-healthy aging people, aging causes structural and functional abnormalities in fat (adipose) tissue. The most readily-observed changes include relative increases in fat tissue in some places (such as the visceral fat that surrounds the gut and liver), even as other places (such as the face) actually lose fat tissue. Meanwhile, fat that is supposed to be stored in those fat cells is increasingly aberrantly stored inside cells of the muscle and liver.
One kind of aberrant cell within aging fat tissue is senescent preadipocytes — the immature precursors of fat cells. Senescent preadipocytes can no which means the same amount of fat per se has to be crammed into a shrinking number of adipocytes, along with aberrant fat storage in muscle and liver cells. All of these changes make the body less able to metabolize glucose, and contribute to abnormal cell function, systemic inflammation, and a rising level of unhealthy free fatty acids circulating in the blood with age.
Additionally, immune cells resident in the visceral fat tissue (especially adipose tissue macrophages (ATMs)) multiply and release signaling molecules that promote inflammation and recruit even more immune cells out of the circulation and into the adipose tissue. Much of this is a short-sighted attempt by the immune system to clean up the wreckage from dying fat cells in the tissue, which happens increasingly with age as the surviving fat cells are increasingly forced to take in too much fat, and as the densifying fat tissue has a harder time providing these cells with an adequate blood supply. All of this makes the visceral fat tissue itself inflamed, which is probably one reason why aged and obese people become insulin resistant.
In obese mice, ATMs also cause a skew in the ratio of different kinds of blood and immune system similar to what is seen in aging, suggesting a possible role for ATMs in this aging change. This may be important, since that same shift in stem cell fate is linked to the rising risk of anemia and blood cancers with age.
Defective T-Cells
T-cells — which include CD4+ (“helper”) and CD8+ (“killer”) T-cells) — are immune cells that specialize in flagging and destroying (respectively) cells that have been hijacked by viruses or by cancer; helper-Ts are also involved indirectly in producing antibodies, and in other immunological functions. T-cells first emerge out of the thymus gland (located behind your breastbone) in a “naïve” state, ready to take on entirely new threats; when as those threats emerge, helper and killer-T cells transmit and receive (respectively) instructions from other immune cells to be on the lookout for the characteristic proteins of the specific threat at hand at the time. After they’ve cleared the body of their target, the body lets most T-cells die off, but a small reserve of memory T-cells are held in reserve, ever ready to be reactivated should their original enemy ever show its “face” again.
The most conspicuous impact of aging on T-cells is the sheer attrition of naïve T-cells, due to some combination of the inability to produce more of them in the degenerating thymus gland (where naïve cells are produced from their precursors) and to aging damage to the lymph nodes, such that those naïve cells that are produced do not receive the signals they need to survive. This leaves behind large numbers of memory T-cells that at least in principle can mount a defense against previously-encountered threats, but shrinking numbers of cells that the immune system can educate and send off to attack threats it has never faced before.
Looking beyond the sheer numbers, aged T-cells become dysfunctional in a variety of ways. The most obvious consequence of the aging of the T-cell system is the slow collapse of immune defenses as people age. Along with other aspects of “immunosenescence,” this is why so many people over the age of 65 die or are hospitalized each winter by viral infections from which younger bounce back after a couple of days in bed; examples range from influenza to West Nile Virus, and the stark reality of COVID-19 as a disease of aging.
Additionally, aging people suffer increasingly with low-level autoimmunity, meaning that rising numbers of T- and B-cells have come to recognize the body’s own, normal proteins as “foreign” and are busy attacking them, leading to tissue damage and chronic inflammation — but not at such a level as to be recognized as a “disease.”
But in the last few years it’s become clear that T-cell dysfunction also contributes to degenerative aging in multiple ways not obviously linked to immune function at all. The most striking recent example of such effects is a T-cell attack on centers in the brain where new neurons are produced, contributing substantially to the loss of neurogenesis with age in mice. The aberrant T-cell populations in aging tissues exhibit a variety of structural and functional abnormalities, but the biology is complex, and different scientific silos unfortunately use inconsistent criteria to define dysfunctional T-cell types. Notably, the cells termed “senescent T-cells” are not the same thing as “senescent cells” in other tissues, and can be stimulated to reverse their abnormal state.
The Rejuvenation Biotechnology Solution
Each of these various kinds of abnormal cells emerge with age in different tissues, and the problems they cause are distinct from one another. Still, the same basic strategy can be used to eliminate the harmful effects of all of them, which is to tear them out by the root by destroying the defective cells themselves. There are two main approaches that may achieve this:
- Develop a drug that is toxic to the unwanted cells, or that makes them commit suicide, but that doesn’t harm healthy, normal cells; or
- Rejuvenate, fortify, or engineer de novo the immune system’s ability to selectively seek out and kill the target cells; or
- Physically remove these cells
Dr. Aubrey de Grey and colleagues argued for the actual destruction of senescent and other abnormal cells in the original SENS manifesto in 2002, at a time when many considered it radical. But opinions changed dramatically after a breakthrough study in transgenic mice in 2011, in which mutant mice line that accumulate very high levels of senescent cells were engineered to carry a genetic “suicide switch” that would be activated when a gene most often expressed in senescent cells was activated. Turning on the “suicide gene” in these mice yielded dramatic effects against aging-like phenotypes in a way that convinced many that the same could be done in otherwise-normal aging mice. That study was followed up with a similar study confirming that ablating senescent cells preserves health and reduces middle-aged deaths in otherwise-healthy aging mice.
These studies were exciting, but still couldn’t be directly used as the basis of rejuvenation biotechnologies that preserve youthful health and function in humans due to the need for the “suicide gene” to be engineered into the mice (or a person). Fortunately, James Kirkland and his coworkers at the Mayo Clinic were already working on a strategy to develop “senolytic” drugs that selectively destroy senescent cells. Kirkland soon showed that giving these senolytic drugs to aging mice delivered rapid-onset rejuvenating benefits . That first report was followed by a flood of additional papers by multiple scientific groups, showing that destroying senescent cells using a wide range of senolytic drug, immune, and gene therapies has sweeping abilities to ameliorate or reverse multiple diseases of aging in animals, from atherosclerosis, to kidney disease, to osteoporosis, to Parkinson’s disease — and beyond.

The same core strategy can be developed into additional therapies that target other abnormal cell types that accumulate in aging people. Remarkably, for example, improves their metabolic health, and reduces the abnormal storage of fat per se in their liver cells. Even more remarkably, the same treatment marginally improves blood sugar metabolism even in young, lean mice, and reduces their need for the hormone insulin to do it. This suggests that a more targeted way to specifically destroy abnormal subsets of ATMs could benefit aging mice — and people.
At the time of this writing (Fall of 2021), Dr. Andrei Gudkov and his team at the Roswell Park Cancer Center are developing a proof-of-concept for future rejuvenation biotechnologies that will ablate cells with active retrotransposon activity, using a transgenic “suicide gene” system similar to the INK-ATTAC system that first demonstrated the rejuvenating effects of destroying senescent cells in aging mammals.
The best way to target defective aging T-cells is less clear, especially as there are multiple different kinds of such cells that the aging body would likely be better off without. One part of the answer might involve the inhibitory receptor PD-1. PD-1 is an immune “checkpoint” that is intended to keep overzealous T-cells attacking from normal, healthy cells in the body. But many populations of abnormal T-cells found in aging bodies also display PD-1 on their surfaces, including the T-cells that inhibit the birth of new neurons in the brain of aging mice; the T-cells in the tumor tissue of many aggressive cancers, which effectively disarms them so they can’t attack the tumor; and other T-cells that accumulate abnormally in aging mice and humans that are likely useless at best and cause destructive immune responses at worst.
So tailored therapies that use PD-1 as part of a targeting system might form the basis for ApoptoSENS therapies targeting aging, dysfunctional T-cells. A promising proof of the general principle came from a study in which scientists destroyed a subset of PD-1-displaying T-cells that accumulates as part of aging in the spleens and visceral fat of mice. In normal-weight mice, the accumulation of these cells with age is dramatically reduced in by anti-aging Calorie restriction. When mice are made obese with a high-fat, high-sugar diet, they accumulate even more of these cells in their fat — and worryingly, this additional burden can’t be reversed by putting them on a diet that largely normalizes their weight. This suggests that aging people, too, may be stuck with these T-cells once they’re established, unless we develop rejuvenation biotechnologies to target them.
As a proof-of-concept for such an approach, scientists used an antibody to destroy all T-cells in late-middle-aged mice that displayed PD-1 on their surfaces. Purging abnormal T-cells from the visceral fat of aging mice lowered their blood sugar and reduced insulin resistance. There were similar effects when such abnormal T-cells were depleted from the visceral fat of obese mice .
In the longer term, implementing the WILT OncoSENS strategy will give us opportunities to make it easier to destroy many kinds of aberrant cells. Should WILT indeed prove to be the only ultimate safe haven against cancer, then part of the process will be the replacement of our stem cells with pristine, re-engineered cells “WILTed” to make them impervious to cancer. The precursors of immune cells like T-cells and ATMs will be one of the easiest and earliest tissues in which to do this. But in the process of engineering these new, cancer-proof stem cells, we could also engineer the replacement cells with “suicide genes” that could destroy aberrant cells upon administration of an activating drug, just as was done in the early mouse studies in the development of senolytic therapies.
No Cell is an Island
The ApoptoSENS strategy is to ablate aberrant cells, but the application of other rejuvenation biotechnologies to the aging body may indirectly cause some such cells to behave themselves, without resorting to the crude messing-with-metabolism approaches that have been the foundation of past attempts at “managing” the diseases of aging and “old school” geroscience.
For example, Belgian scientists have shown that the abnormalities in ATMs in the visceral adipose tissue of aging mice are in significant part caused by the depletion with age of a kind of regulatory eosinophil immune cell. Repleting aging adipose with young eosinophils “tames” the aging ATMs, yielding wide systemic benefits like more youthful strength, endurance, and immune response to a vaccine-like injection. So in this case, a RepleniSENS strategy might obviate or greatly reduce the need for ApoptoSENS approaches against these cells.

Similarly, two groups of geroscience researchers have shown that the SASP from accumulating senescent cells in the visceral fat of aging mice sounds an alarm that sends ATM precursors rushing into the tissue. Once recruited, the SASP from senescent cells stimulates these aggravated ATMs to express CD38, an enzyme that generates inflammation while eating up NAD+. NAD+ is a critical energy-carrier molecule that declines in our cells with age — a potential problem, since NAD+ is a necessary cofactor for many cellular processes, including regulation of gene expression and one mechanism of DNA repair. Inflamed and depleted of NAD+, aging adipose tissue becomes metabolically dysfunctional. Periodically ablating senescent cells in mice starting in middle age prevented the age-related rise in CD38 expression in adipose tissue, and partially normalized tissue NAD+. Thus, ApoptoSENS against one aberrant cell type (senescent cells) reduced the need for a separate ApoptoSENS therapy to target ATMs.
Where We Are Now
The exciting effects of senolytic drugs in animal studies has sparked an unprecedented surge in research, resulting in the launch of multiple senolytic biotech startups, with companies and capitalists placing their bets on a wide range of different strategies to destroy these foot-dragging renegades. While most senolytics are still in preclinical development, there are a substantial number of early-stage clinical trials of senolytic therapies underway. In these trials, senolytic drugs are being administered to people suffering with a variety of diseases of aging that are strongly linked to senescent cells, from osteoarthritis to COVID-19 to Alzheimer’s. A couple of these trials have been completed — but while the results have been suggestive ofsuggest benefit, they’re such small, early-stage studies that they can really only show us that the tested senolytics safe enough in the very short term. The next step will be to proceed with the necessary larger, longer-term trials that will tell us with confidence which of these diseases can be effectively targeted with senolytic drugs.
Unfortunately, there has been big setback at UNITY Biotechnology, which is one of the leaders in the senolytic space. A trial of their senolytic candidate for osteoarthritis failed to meet its clinical endpoint. This reminds us that the progress of science and medicine is littered with good ideas that fail and timelines that aren’t met — but equally, that the failure of this one senolytic for this one indication shouldn’t be over-extrapolated to the rest of the senolytic space. Consider the history of statins and other medicines to lower LDL (atherogenic) cholesterol (LDL-C). Statins are today some of the most widely-used drugs in the developed world, and have prevented untold numbers of heart attacks, strokes, and early deaths — but before the breakthrough with statins, multiple LDL-C-lowering drugs failed in clinical trials, including some candidates that actually increased heart attacks or deaths. But after the success of lovastatin — the first, critical drug in the statin class — a whole field broke open. Lowering LDL-C via diet or any of multiple drugs with different mechanisms of action is now the mainstay of the prevention and treatment of atherosclerotic cardiovascular disease, and is well-established to lower major adverse cardiovascular events (MACE) — and in the case of statins, to increase survival.
Fortunately, UNITY have climbed back in the saddle and are pressing on, targeting senescent cell elimination with different senolytics for diseases of the aging eye, and have announced promising initial results in an early-phase clinical trial for patients with these indications.
A key technology that would accelerate progress in turning the science of senolytics into working human rejuvenation biotechnologies would be a simple, noninvasive biomarker of senescent cell burden. It’s easy enough to show that a senolytic drug eliminates large numbers of senescent cells in animals, but the methods are of no use for testing them in humans: they involve either killing the animals, or invasive biopsies, or engineering transgenic animals from birth such that cells expressing one important regulator of senescence light up under light of the right frequency. So scientists and startups are today are using senolytic therapies that work brilliantly in mice, but are in large part flying blind once they start using them in humans.
A reliable, noninvasive biomarker of senescent cell burden would therefore unlock the potential of the the senolytic field, because it would give scientists, startups, and investors more confidence in even early-stage trials that candidate senolytic drugs could actually destroy substantial numbers of senescent cells in living humans, as opposed to in laboratory mice or human cells in a Petri dish. Being able to validate “target engagement” this way would facilitate clinical trials of senolytics, prevent costly late-stage failures, and de-risk companies for investors, encouraging more funding for companies with viable senolytic candidates.
One potential such biomarker that has been demonstrated in mice is to measure blood levels of a lipid signaling molecule called dihomo-15d-PGJ2. This molecule is normally only found inside of senescent cells, but when senescent cells are destroyed with senolytic drugs, the cell bursts and dihomo-15d-PGJ2 escapes into the circulation where it can be measured. So monitoring levels of dihomo-15d-PGJ2 might be used as a biomarker of successful senolysis rather than of senescent cell burden per se.

SENS Research Foundation Research
In the early days, SRF funded work by Kevin Perrott in Dr. Judith Campisi’s lab at the Buck Institute, screening libraries of natural compounds for potential senolytics (as they would later be known). No senolytics were identified, but they did discover that the phenolic compound apigenin inhibits the SASP in senescent cells in vitro. This was one of an ongoing series of projects SRF has funded in Dr. Campisi’s lab.
The Foundation also sponsored projects aimed at developing f a T-cell scrubber that provided some early proof-of-principle for the idea of clearing specific classes of dysfunctional T-cells or other cells in the circulation, along with a pilot animal study to simulate the effects of combining such a purge with stimulating the thymus gland to produce more fresh, naïve T-cells.
We also made a Mission-aligned investment in Oisín Biotechnology, a company developing a kind of temporary “suicide-gene therapy” targeting senescent cells the same way they had been targeted in the first animal lifespan studies.
Currently, SRF is working on several approaches to better target senescent cells. One such project is to rejuvenate, transiently enhance, or supplement the elimination of senescent cells by the innate immune system. Another is to better target secondary senescent cells — the subpopulation of senescent cells that emerge when SASP factors from initially-triggered senescent cells force neighboring healthy cells to become senescent, setting off a chain reaction whose termination our preliminary data suggests will require novel rejuvenation biotechnologies.
Additionally, SRF is breaking open an entirely new apoptoSENS field: the ablation of cells with activated retrotransposons. With SRF funding, the Gudkov Lab is engineering mice with a transgenic “suicide gene” system built into their cells, similar to the INK-ATTAC system that first demonstrated the rejuvenating effects of destroying senescent cells in aging mice. But in this case, the suicide gene system will instead be triggered by the initiation of interferon signaling that often (but not only) occurs after retroviral reactivation. Just as the INK-ATTAC mouse paved the way to the development of today’s senolytic therapies, this suicide gene system for the elimination of cells harboring reactivated retrotransposons holds the promise of paving the way for similarly-powerful future “retrolytic” rejuvenation biotechnologies.
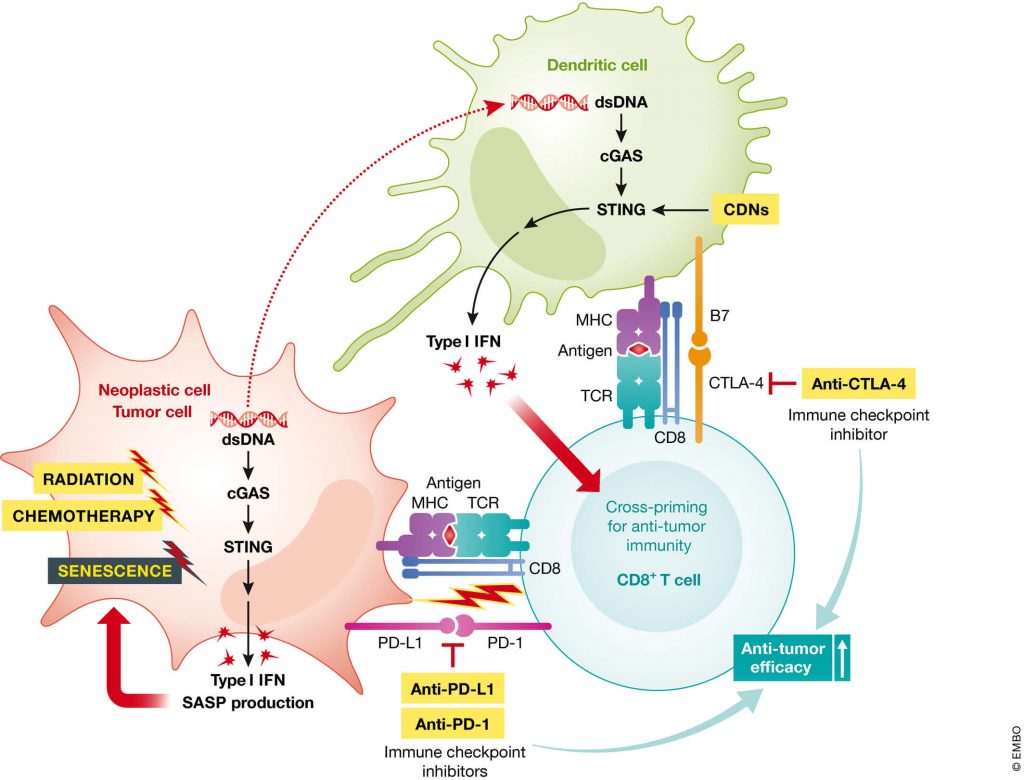
Work on this “suicide gene” system for reactivated retrotransposons began several years ago, with Dr. Gudkov’s lab constructing the system and inserting it into cells. Unfortunately, his team has run up against roadblocks twice in trying to build a version of the system into the cells of mice. His lab has requested funding to be held in reserve for work on a new mouse model as soon as Dr. Gudkov figures out a more viable path to success on this front.
Meanwhile, in 2021, we are funding a new project testing combination longevity therapeutics (a neglected but critical gap in the rejuvenation research agenda) — in this case, applying a form of RepleniSENS (stem cell transplant) after a round of ApoptoSENS therapy (senolytic drugs to ablate senescent cells), each of which have been shown on their own to benefit healthspan and median life expectancy, without actually breaking the lifespan barrier.