
A range of damaged proteinaceous aggregates accumulate intracellularly and extracellularly in the aging brain, with higher burdens of characteristic aggregates associated with diagnosed age-related neurodegenerative disorders. These include beta-amyloid protein (Aβ) and neurofibrillary tangles (NFT) in Alzheimer’s disease (AD) and other age-related dementias, and Lewy bodies and other intracellular α-synuclein (AS) aggregates in Parkinson’s disease (PD) and other synucleinopathies. Additionally, it is increasingly clear that LB along with other neuronal protein aggregates are key drivers of “normal” cognitive aging.
Multiple lines of evidence — from cell culture studies, transgenic model organisms, and genetic epidmiology — link a person’s steady-state AS levels and accumulation of LB to both clinical PD and subclinical age-related movement disorders. Mutations and genetic variants that increase the production or aggregability of AS are clearly linked to earlier onset and severity of PD. Three different point mutations in the SNCA gene that encodes the AS protein are causal for dominantly-inherited disease PD.[1-3] So are duplications[4] and triplications[5,6] of the wild-type (WT) SNCA allele. In such cases, the gene dose correlates with the severity of disease,[7,8] and with blood levels and cerebral cortex deposition of AS. Notably, the relative increase in deposition of aggregated AS in these cases is disproportionate to the chronic increase in soluble AS levels.[9] These SNCA mutations, as well as more common SNCA polymorphisms,[10-13] are more robustly associated with higher risk of diagnosed PD and other synucleinopathies than any other disease-associated genetic variants. Some of the common disease-associated SNCA variants associated with “sporadic” PD also increase gene expression[11] or translation and AS protein accumulation,[13] again consistent with a dose-response relationship.
Even absent disease-associated genetic variations, AS levels rise with age in the dopaminergic (DA) neurons of the substantia nigra pars compacta (SNc), whose loss with age and toxicity is central to the classical motor symptoms of PD.[27] Animal models overexpressing WT human AS develop motion disorders that particularly resemble the extradopaminergic symptoms of PD,[21,22] and human disease-associated transgenes cause more severe neuropathology and behavioral abnormalities.[23,24] Mutations that increase the production of soluble prefibrillar/oligomeric AS aggregates are neurotoxic to cells in culture, and several means of blocking or reducing aggregation reduce or eliminate this toxicity: these include protein chaperones, deletion or disruption of the mutant proteins’ aggregation-prone hydrophobic domains, or even promoting the assembly of short protofibrils into higher-order assemblies.[23]
During the biological aging process, LB proper first appear in the peripheral and autonomic nervous systems (including spinal, cardiac, vagal, enteric, and genitourinary neurons), along with the olfactory bulb and lower brainstem. At this early stage in progression, subjects who go on to develop PD do not yet exhibit the characteristic motor deficits of the disease, but are often troubled by a range of more subtle extranigral symptoms, including urinary urgency, constipation, pain, orthostatic hypotension, and executive dysfunction.[14-20] As the disease process continues, LB spread caudally through the substantia nigra, areas of the midbrain and basal forebrain, and finally the neocortex (Figure 1).[14,15,17] More proximate to the emergence of classical motor symptoms, hyposmia, rapid eye movement (REM) sleep behavior disorder (RBD), and cardiac sympathetic dysfunction become more common, specific, and predictive symptoms.[14,20]
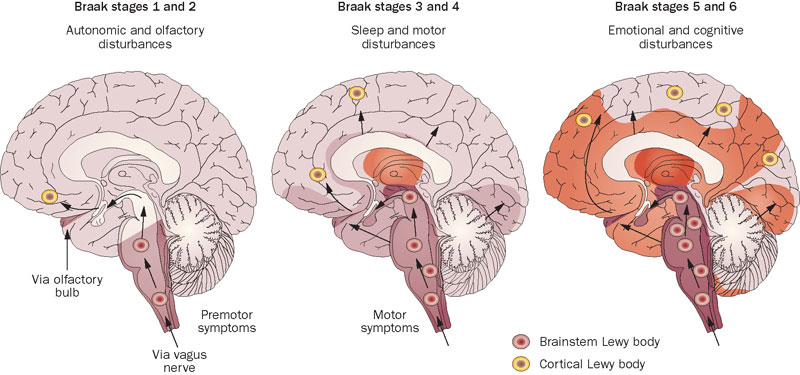
Figure 1. Spread of α-Synuclein in the Brain. Reprinted from [25,26]. With permission, Nature Publications Group.
A Warning Sign on the Road Ahead
Granted the strong connection between AS levels, LB accumulation, and risk of PD within the bounds of current life expectancy, one potential therapeutic strategy — consistent with twentieth century medicine’s risk factor “management” heuristic — would be to develop drugs that inhibit the production of AS protein. But native, non-aggregated AS is a physiological protein, whose functions remain little understood but which likely involve the regulation of intersynaptic vesicle mobility, the vesicle recycling pool, and neurotransmitter release.[23]. Suppressing the production of AS necessarily entails interfering with its homeostatic regulation and its physiological functions.
Two recent studies foreshadow the risks that accompany this strategy. One was a genomic epidemiological study on the course of PD in 1,098 cases, tested for their SNCA promoter allele REP1 genotype.[28] As compared to the WT (261 bp) REP1 allele, longer (263 bp) variants increase SNCA gene expression, and have been associated in previous studies with increased risk of “sporadic” PD, while carriers of shorter alleles (259 bp) have lower gene expression and reduced PD risk.[11] Additionally, it had previously been reported that among persons suffering with idiopathic PD, carriers of the 263bp variant are four times as likely to suffer rapid motor decline (≥5 point increase in the annual rate of change in the Unified Parkinson’s Disease Rating Scale motor score) as were noncarriers with the disease.[29] That study also found a trend toward lower risk of rapid motor decline in carriers of the short (259 bp) variant.[29] Yet in the new report, carriers of the short variant with established PD fared worse than noncarriers on both motor and cognitive outcomes.[28]
If the new study’s findings are confirmed, it would imply that the short SNCA variant — and perhaps steady-state physiological AS levels — exert paradoxical, antagonistic effects across the course of PD.[28] Low native AS levels may delay the onset of PD, presumably through decreasing the sporadic formation of AS oligomers and/or LB in the brain and peripheral neurons. Yet once the disease is established, lower production of AS may be harmful, perhaps because of loss of physiological AS functions when low production is combined with sequestering of native protein into nucleating AS aggregates in disease-affected neurons. As the authors conclude, “Our results raise the possibility of a dual and opposing role of SNCA expression in pre-clinical versus clinical PD (“Janus effect”). This raises concerns about the efficacy and safety of therapies aiming to reduce SNCA expression in PD.”
A second study[30] raises the wider specter of unanticipated consequences of suppressing production of a native, disease-associated protein. A significant body of evidence has accumulated that native tau protein acts as an effector of much of the neurological dysfunction associated with Aβ in AD. In particular, reduction of expression of native tau protein in mice transgenic for mutant forms of amyloid precursor protein (APP) that are determinative for early-onset AD is protective against cognitive deficits.[31-35] The authors of the study under discussion[30] had noted a substantial body of evidence suggesting that tau might also have a pathological relationship with AS, so that a reduction in endogenous tau expression might be similarly protective against AS-driven motor and/or cognitive dysfunction:
AD and PD overlap clinically. A proportion of patients show manifestations of both diseases [citation]–[citation], and first-degree relatives of patients with early-onset AD are at increased risk of developing PD [citation]. The two diseases also overlap pathologically. A substantial proportion of AD patients have Lewy bodies in their brain … [citation]–[citation]. Conversely, a proportion of PD patients have amyloid plaques … [citation], [citation] … [and] hyperphosphorylated tau [citation]–[citation], which aggregates in AD and in some cases of PD without dementia [citation]–[citation]. In addition, specific variants of the human tau (MAPT) gene appear to be genetic risk factors for PD [citation]–[citation] … Furthermore, neuronal overexpression of human α-synuclein in transgenic mice causes phosphorylation and aggregation of endogenous tau [citation], [citation]–[citation] [and cf. [36,37] below]. In light of these findings and a recent report that tau reduction prevented dendritic degeneration in a neuronal culture model of mutant LRRK2-linked PD,[[38] below] we wondered whether tau reduction is also beneficial in mouse models of PD.[30]
To test this hypothesis, the investigators imposed either of two models of PD on mice with Tau+/+, Tau+/–, or Tau–/– mouse backgrounds. Despite the strength of the evidence on which the experiment was based, reduction in endogenous tau levels was not beneficial in either PD model. In fact, tau-reduced mice appeared more vulnerable to unilateral striatal injection of the selective dopaminergic neurotoxin 6-hydroxydopamine (6-OHDA), only mice, since they exhibited increased latency on the pole-descending test as a result and WT mice did not.[30]
Moreover, it would later be reported that knocking out native tau in otherwise WT mice induces profound motor and cognitive deficits reminiscent of Parkinsonian-dementia disease, associated with SNc dopaminergic neuron loss.[39] The mechanism that apparently underlay these neurotoxic effects was quite unexpected: the investigators uncovered a previously-unknown role for tau in APP-mediated iron efflux, which was impaired by tau knockout. Importantly, the neurotoxic effects of tau knockout took 12 mo to emerge in the mice, who had behaved indistinguishably from WT at 7 mo of age.[39] One possible way to reconcile these findings with the previous reports of protection afforded by reducing endogenous tau levels against the cognitive deficits imposed by AD-associated APP mutations[31-35] may be a time-dependent antagonistic-pleiotropic effect similar to that posited for the SNCA length polymorphisms,[11,28,29] since the newer study in WT mice was longer-lasting and the overproduction of aggregate-prone mutant APP in the AD model mice is so severe.
Such results[30,38,39] emphasize the profundity of our ignorance about the complex metabolic roles of these disease-associated proteins in their native conformation, and thus the parlousness of interventions based suppressing their production — particularly in so precious an organ as the brain. Instead, the regenerative engineering approach to the prevention and reversal of age-related neurodegenerative disease arising from AS and tau aggregates rests not on suppressing the production of their native protein precursors, but on therapeutic clearance of the aggregates themselves. In this “damage-repair” heuristic, APP, AS, and tau are left under physiological regulation, unperturbed so long as they maintain their native conformation and therefore able to continue carrying on their essential physiological functions. Instead, rejuvenation biotechnologies target only disease-associated aggregates — aggregates that are no longer able to carry out their physiological function, but exert an entirely deleterious influence on the cell, whether mechanically, metabolically, or biochemically. This approach is conceptually attractive because it does not perturb normal metabolic function, but only returns biological structures to their youthful default state. And from restored biological structure flows restored biological function.[78-80]
Immunotherapy Against Age-Related Neurological Disorders
The use of immunotherapy to remove extracellular aggregates from the aging brain is an extremely active field of Alzheimer’s research, with multiple active and passive Aβ vaccines currently in human clinical trials.[40] The most advanced of these is solanezumab/LY2062430 (Eli Lilly), which is one of three Aβ antibodies that are presently entering into late-stage trials in preclinical or early-stage AD.* The main immunotherapeutic approaches can be divided into active vaccination (in which the target aggregate or a fragment thereof is injected into patients in order to elicit the generation of therapeutic antibodies endogenously) and passive vaccination (using infused monoclonal antibodies targeting the aggregate). Additionally, numerous Aβ immunotherapy agents are advancing along the clinical pipeline from preclinical studies, using strategies such as Aβ-targeting affibodies, DNA and peptide vaccines targeting beta-amyloid epitopes, and infusion of catalytic Aβ-cleaving IgM antibodies instead of IgG-based binding antibodies (this last approach being the most conceptually exciting of all — see Figure 2).
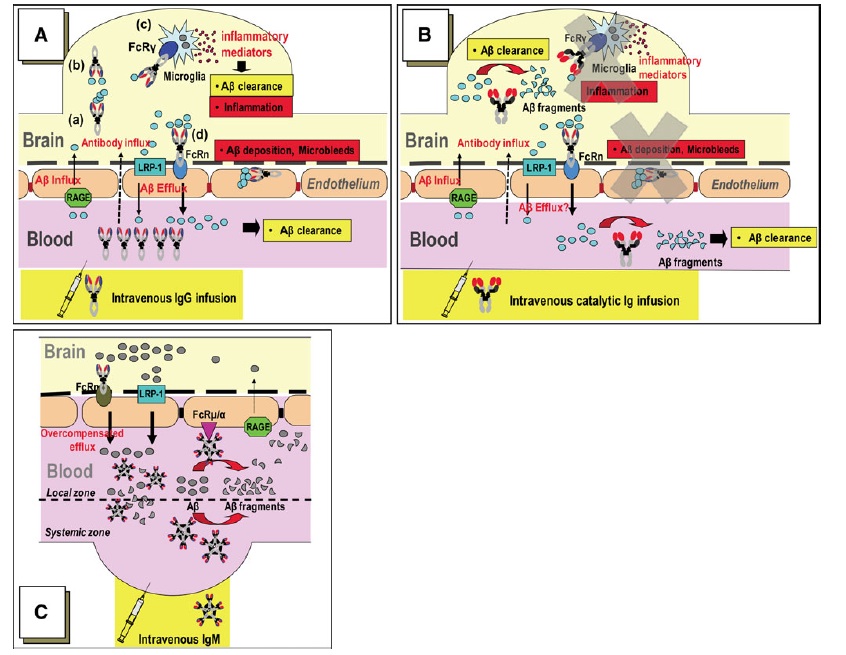
Figure 2. Comparison of Mechanisms of Aβ Clearance by Immunotherapeutic Approaches. (A): With IgG-based “capturing” immunotherapies, antibody-bound Aβ is taken up by microglial Fcγ receptors and degraded in the lysosome, and/or IgG-bound Aβ may be taken up by neonatal Fc receptors (FcRn) at the BBB, leading to BBB transcytosis and peripheral efflux. The former is likely involved in the inflammation associated with active vaccination with AN1792; the latter may contribute to vasogenic edema following deposition of stable complexes in the cerebral vasculature. (B): catalytic IgM or IgM-derived IgV clones should abrogate these potentially deleterious effects. Instead, Sudhir Paul’s group hypothesizes that (C) Aβ hydrolysis in the periphery increases the trans-BBB Aβ concentration gradient, while IgM binding to BBB Fcμ/α receptors promote direct Aβ transcytosis by FcRn. Reproduced from (59), with permission from Springer Verlag and Dr. Paul; © Springer Verlag, 2010.
But as we have reviewed in several previous blog posts on this subject, immunotherapeutic approaches are now also being used to target intracellular aggregates, including pathological tau species. Perhaps surprisingly, these studies have reported that antibodies raised against tau or particular disease-associated epitopes does indeed reduce the burden of these aggregates, despite their intracellular localization, and that this is accompanied by prevention or alleviation of behavioral deficits. Prior to this work, Gunnar Gouras and coworkers had shown that some Aβ-targeting antibodies may enter the cell and clear intraneuronal Aβ, and the mechanisms for such effects continue to be the subject of research.[41] Additionally, a mechanism involving blocking the trans-synaptic propagation of “infectious” pathological tau species by binding them in the synaptic cleft has been postulated[42] and received some experimental support.[43] A recent article in the scientific references Michael Hutton of Eli Lilly for the estimate that close to a dozen companies (including Lilly itself) are pursuing tau immunotherapy.
Preclinical studies also provide proof-of-concept for AS vaccination. In an early study,[52] Eliezer Masliah and colleagues at the University of California at San Diego collaborated with Dale Schenk (then of Élan Pharmaceuticals; now of Prothena Corp PLC) to test the effects of active AS vaccination in transgenic mice expressing WT human AS (a model of PD, or arguably dementia with Lewy bodies (DLB). Active vaccination of these animals with with the recombinant human WT AS reduced the accumulation of AS inclusions and oligomers in neurons and presynaptic terminals in these animals, and simultaneously curtailed the associated synaptic degeneration.[52] Notably, the effects of vaccination were more pronounced in animals who produced antibodies with higher (vs. lower) affinity for their target in response to vaccination. Additionally, the authors presented evidence that the elicited therapeutic antibodies were taken up by neurons, localized with lysosomes and intraneuronal AS inclusions, and increased the levels of the lysosomal protease cathepsin D.[52]
Several years later, Masliah, Schenk, and colleagues again collaborated, this time in a passive vaccination study in the same mouse model.[53] From Élan’s panel of monoclonal antibodies specific to either the N- or C-terminus of human AS, the team identified 9E4, which targets the C-terminus, as the most specific. Transgenic human AS mice in their study suffered the loss of ≈55% of their postsynaptic densities and a reduction of ≈25% in presynaptic terminal diameter; immunotherapy with 9E4 substantially rescued this damage. The PD mice also exhibited deficits in learning and memory on the water maze test; in motor coordination on the rotarod; and in motor function on the pole test. Passive vaccination with 9E4 normalized performance on the rotarod (see Figure 3 C), and improved performance on the pole test (Fig. 3B) and nearly abrogated most measures of impaired performance in the water maze. Treatment also attenuated the accumulation of putatively key neurotoxic calpain-cleaved AS aggregates in neuronal cell bodies and neuropils of the temporal cortex and hippocampus, and the associated neurodegenerative deficits.[53]
The study also showed that labeled 9E4 antibodies are trafficked into the CNS and internalized by AS-containing neurons of PD mice in vivo, where they subsequently colocalize with lysosomes and autophagosomes.[53] Moreover, tissue sections from vaccinated animals exhibited increased colocalization of labeled AS in granular structures positive for the autophagy marker and effector microtubule-associated protein light chain 3.[53] Combined, these data again support a model in which AS-targeting antibodies traverse across the blood-brain barrier, bind synaptic or membrane-associated AS, are taken up by neurons with their captured AS, and transport AS to the lysosome for autophagic clearance, as was suggested in their prior work with active AS vaccination.[52]
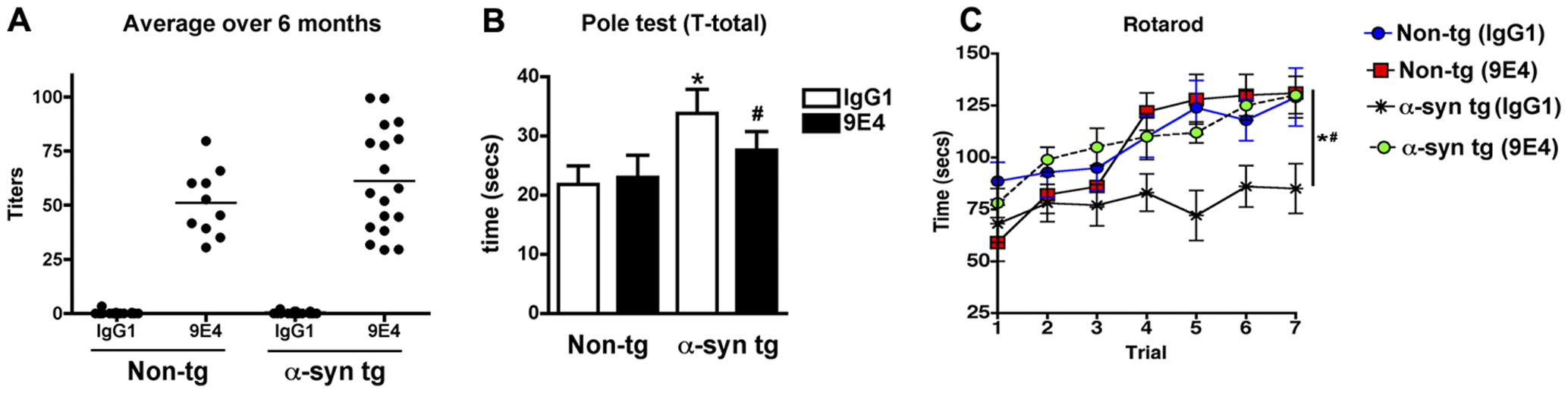
Figure 3. Passive AS Vaccine Alleviates PD-Like Phenotypes. Circulating antibodies (a) and pole test (B) and rotarod (C) performance in transgenic mice expressing human wild-type AS after passive vaccination with AS-targeting antibody 9E4 or non-immune control IgG1. Reproduced from [53] under Creative Commons License from PLoS ONE.
In addition to their effects on neurons, there is evidence that at least some AS immunotherapies additionally clear synaptic AS aggregates by increasing the microglial uptake and degradation of synaptic AS aggregates. In a subsequent report, Masliah and coworkers showed that two out of four tested AS-targeting monoclonal antibodies promoted the uptake of AS aggregates by microglia in vitro. The more effective of these was an antibody specific for the C-terminus of human AS, termed antibody 274 (Ab274), which was then the subject of further investigation. The results demonstrated that antibody-bound AS aggregates were taken up by microglia (but not astrocytes or neurons) via Fcγ receptors, and then trafficked to the lysosome via lipid raft-mediated endocytosis. This led to reductions in AS aggregates but not monomers in the medium.[60]
Consistent with these cell culture results, injection of Ab274 into the hippocampus of transgenic PD/DLB model mice increased microglial activation and the number of microglia staining positive for human AS on the ipsilateral side, whilst reducing astroglial and neuronal AS accumulation. And similarly to their previous studies with 9E4, treatment with Ab274 normalized performance on the pole test and prevented the increased hyperactivity in seen in TG animals treated with non-immune control IgG in the open field test (as compared with WT mice treated with either control antibody or Ab274). Transgenic AS mice treated control IgG also suffered a >30% loss of hippocampal neurons relative to WT mice treated with either antibody, which was abrogated in transgenic animals receiving Ab274; a similar 28% deficit in synaptic counts (as quantified with synaptophysin staining) was substantially ameliorated by Ab274 treatment.[60]
Buoyed by the strong evidence from tau and AS vaccination studies, an Austrian biotechnology firm with an unique development platform — with support from a major Parkinson’s charity — has lept ahead of the pack. As of this writing, they are now in the midst of human clinical trials of a first-in-class immunotherapeutic agent targeting the removal of pathological AS species as a disease-modifying rejuvenation therapy to prevent, arrest, and reverse the ravages of Parkinson’s disease.
AFFITOMEs: Molecular Mimicry for Innovative Vaccine Design
Founded in 2003, AFFiRiS AG was established to exploit its patented “AFFITOME” technology, an innovative method of creating tailor-made peptide-mimetic vaccines that in principle should sidestep many of the difficulties that have mired past Aβ immunotherapy candidates. Existing immunotherapies targeting the removal of pathological aggregated proteins all involve the use of antibodies that directly target the sequence and/or conformation of the offending aggregate. Active vaccines do so by injecting patients directly with the offending aggregate, triggering the patient’s immune cells to raise antibodies against it; passive vaccines do so by first vaccinating mice (instead of human patients) with the target aggregate, and then humanizing their protein sequences before scaling them up via recombinant DNA constructs to generate monoclonal antibodies that can be infused preformed into human patients.
Each of these approaches have their limitations. Active vaccine approaches entail the risk that, along with the necessary B-cell response, exposure to the target aggregate may also result in an autoreactive T-cell response against the it, leading to the possibility of an inflammatory attack centralized on the aggregate’s cerebral deposition sites. Just such a Th1 immune response is postulated to have underlay the meningoencephalitis that befell some 6% of patients in the Phase II trial of the original AN1792 Aβ vaccine, leading ultimately to its discontinuation.[44] Indeed, autoimmunity has emerged in preclinical work as a potential risk for active AS vaccination, in an animal study using a nitrated AS species typical of LB as the vaccinating agent.[62]
Passive vaccines eliminate the risk of an autoreactive T-cell response by bypassing the patient’s immune system altogether, generating the therapeutic antibodies from without and infusing them into the patient. However, it still runs some risk of off-target effects if the resulting antibodies bind to similar epitopes on related proteins. In the case of immunotherapies targeting AS, one such potential iatrogenic bystander is β-synuclein, which has a similar expression pattern and sequence homology to AS and yet is not found in LB, is not amyloidogenic, and indeed appears to have neuroprotective effects by inhibiting AS aggregation and other mechanisms.[45,46]
The AFFITOME system aims to avoid both these risks by exploiting molecular mimicry in the design of novel antigens for active vaccines: the tendency of the immune system to cross-react from a primary target antigen to other, unrelated peptides (“neo-epitopes”) with shared amino acid sequences and/or similar conformational structures. The first step in designing new vaccine antigens with the AFFITOME system is to create a kind of positive control, termed a selecting antibody, against which candidate vaccine peptides will be tested for their ability to immunologically mimick the behavior of the target antigen. Selecting antibodies are generated by injecting mice with the original target aggregate or a key peptide sequence in order to induce the production of binding (IgG-type) antibodies. In passive vaccine approaches, these antibodies would be humanized and turned into biologic therapeutic; in the AFFITOME system, they are instead used to screen the ability of candidate neo-epitope vaccines (“AFFITOPEs”) to exhibit molecular mimickry for the candidate antigen by engaging antibodies raised against the target antigen itself.[47,48]
The fact that candidate AFFITOPEs are peptides with amino acid sequences unrelated to the original target antigen should in principle significantly reduce the risk of cross-reactive autoimmunity against off-target physiological proteins. Ideally, AFFITOPEs would not contain immunogenic sequences in common with any physiologic protein; at minimum, they can be restricted to avoid cross-reactivity with particular peptide sequences of a priori concern. In AFFiRiS’ PD pipeline, this included exclusion of sequences present in β-synuclein, out of concern for its structural similarity to AS.[45,46,49,50]
Conversely, the fact that neo-epitopes lack sequence identity with actual human proteins (and are therefore immunologically “non-self”) should reduce the induction of immunological self-tolerance to the elicited antibodies[54] which would otherwise be expected to impede a robust immunological response to vaccination with an antigen containing “self” sequences such as AS itself.[47]
Beyond the broad properties expected to be inherent to neo-epitope vaccines, the use of neo-epitopes rather than primary target antigens as the immunizing agent is proposed to give the AFFITOME system more flexibility in creating a wide pool of potential therapeutics and then narrowing that pool based on predefined criteria identified a priori as important for safety and/or efficacy. In their AD vaccine pipeline, for instance, AFFiRiS scientists excluded candidate AFFITOPEs with close sequence similarity to the central or carboxy terminus region of the Aβ peptide, based on evidence that the critical sequence responsible for the autoreactive T-cell response to AN1792 was located in these regions.[48,51]
An additional such criterion is sequence length. CD8+ T-cells are activated by peptides 8-9 amino acids in length that are presented by MHC class I, and CD4+ cells by even longer sequences. And a third such criterion is the absence of particular anchor residues in the peptide’s sequence. Thus, by restricting the pool of candidate AFFITOPEs to peptides of less than 8 amino acids’ length, and rejecting those with specific amino acid sequences in anchor positions, specific T-cell reactions are expected to be abrogated.[47,48]
On the other hand, an effective AFFITOPE vaccine must still engage B-cells to elicit a humoral immune response and elicit therapeutic antibody production upon injection into patients. This introduces an apparent dilemma in neo-epitope sequence length criteria: to activate humoral immunity against an antigen, a vaccine must activate CD4+ T-helper cells, which requires the presence of epitopes of 14-20 amino acids in length bound to MHC class II molecules upon presentation. Yet as discussed above, the active aggregate-targeting neo-epitope must be restricted to peptides of <8 amino acids in length to avoid induction of autoreactive T-cells. In the case of PD101, this circle has been squared by conjugating the primary neo-epitope with a carrier peptide (in this case, the workhorse keyhole limpet hemocyanin) with suitable T-helper epitopes but no sequence homology with human proteins.[49] To some extent, the peptide pool can even be further tailored from prior knowledge: in the case of AD, this may include the fact that the B-cell epitope for Aβ is located within the first 11–15 amino acids of its N-terminus.[51]
The intrinsic and designed safety features of the overall AFFITOME system for generating candidate AFFITOPEs make AFFiRiS comfortable with the use of aluminium hydroxide as an adjuvant in their vaccines, despite what many believe is its strong tendency to induce or exacerbate autoimmune reactions.[55] Combined, all of these features would be expected to lead to the creation of vaccines that elicit powerful, precise immunological responses against their pathological targets, with little risk of deleterious immunological consequences.[47] In tentative support of the expectation of safety built into the selection process of the AFFITOME system, there were no autoimmune reactions or indications of myeloencephalitis in any of the 48 patients administered AFFiRiS’ Aβ-targeting AD01 and AD02 vaccines in Phase I trials.[47]
In the AFFITOME system, candidate AFFITOPE vaccine antigens are first screened for their ability to be bound by selecting antibodies for the primary target, to give an indication of immunogenic molecular mimicry. Candidates that survive this initial screen can then be tested in vivo — first by evaluating the humoral immune response of mice injected with candidate AFFITOPEs, and then by proof-of-concept studies in animal models of disease (see Figure 4).[48] From there, candidate AFFITOPEs can be taken on to clinical trials.
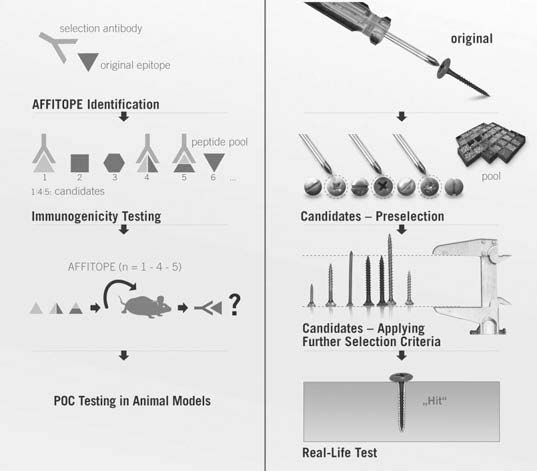
Figure 4. The design algorithm for AFFITOPE vaccines. Reproduced from [48]. ©2009 Springer Verlag.
So far, the AFFITOME system seems to be productive. AFFiRiS completed Phase I studies with its Aβ-targeting AD01 and AD02 vaccines in 2008, with good safety findings for both vaccines; they have an ongoing Phase II trial of AD02 in ≈300 subjects with early AD; and they completed Phase I safety study of a newer AFFITOPE vaccine (AD03/MimoVax) that targets N-terminally truncated and pyroglutamate-modified Aβ species.[47] While no results have been published, the MimoVax Consortium website affirms that MimoVax “met its primary endpoint … and successfully passed phase I clinical testing.” AFFiRiS is now enrolling patients from the original Phase I MimoVax trial in a followup study to evaluate MimoVax’s longer-term safety.
All of this would already be positive news, adding yet more contenders to the hot race for immunotherapeutic rejuvenation biotechnologies targeting Aβ for the arrest and prevention of Alzheimer’s disease (and, eventually, “normal” cognitive aging). But as alluded above, beyond joining the pack in pursuit of Aβ vaccines, AFFiRiS has now broken truly new ground, using its AFFITOME technology to develop an AS AFFITOPE and for the first time advance AS immunotherapy into human clinical trials.
PD01: an AFFITOPE Vaccine Targeting α-Synuclein
It is not clear exactly when AFFiRiS began work on an AFFITOPE vaccine targeting AS, but in March of 2010 the company announced
an important step forward in the development of its vaccine for Parkinson’s disease. The vaccine, known as PD01, has undergone numerous preclinical tests which have confirmed its principle of action (“Proof of Concept”). … Dr Walter Schmidt, director and co-founder of AFFiRiS AG stated: “We have had PD01 evaluated in various preclinical systems. All the tests have shown that this vaccine works against alpha-syn. In addition to histological and biochemical analyses, we have even been able to observe behavioural improvements as a result of our vaccination.” … The company is now starting to prepare for clinical trials of the vaccine which are planned to commence at the beginning of 2011.
In a subsequently-published review article, AFFiRis scientists affirmed that the “AFFiRiS PD drug development program focuses on AFFITOPES eliciting Abs recognizing α-syn while sparing β-syn. It delivered a series of candidates. One AFFITOPE, termed PD01, was found to exhibit disease-modifying activity in POC studies in animal models of the disease.”[47]
Despite these statements, it appears that further preclinical testing was necessary, as the following month a second AFFiRiS press release announced that they had sought out and received a $475,000 grant from the Michael J. Fox Foundation for Parkinson’s Research (MJFF) to extend preclinical testing. In their own page for this grant, MJFF elaborated:
we will be further evaluating the effects of PD01 within a defined pre-clinical screening program … includ[ing] the assessment of immunogenicity, stability and safety as well as efficacy of this vaccine to reduce PD-like pathology in the models used. The effects detectable will be compared to previous results obtained with PD01 at AFFiRiS in order to get additional efficacy and safety information … [and] further elucidate the mechanism of action …
Although the results of these further studies in animal models remain unpublished, they were evidently successful. A professional press article in 2012 reported that “Unpublished experiments performed in collaboration with Eliezer Masliah … demonstrated that … PD01 reduced the levels of alpha-synuclein in the brain, prevented neuronal loss and led to cognitive improvements in two transgenic mouse models of Parkinson’s.”[57] In a contemporaneous review on “Vaccination for Parkinson’s Disease,”[49] AFFiRiS scientists stated that
Preclinical studies involving the subcutaneous administration of AFFITOPE® PD01 demonstrated the induction of a humoral immune response fulfilling the prespecified criteria: reactivity towards a[lpha]Syn[uclein] [see Figure 5 below], but not b[eta]Syn[uclein] … AFFITOPE® PD01 reduces the level of cerebral aSyn and ameliorates aSyn-triggered neuropathologic alterations such as neuronal cell loss and dendritic density in two independent mouse models of synucleinopathies. Most importantly, immunization with AFFITOPE® PD01 was found to translate into a functional benefit as well. Compared to controls, PD01 treated animals show for example superior cognitive functions as assessed by the Morris water maze test.”
And a story in the professional press reports on PD01 data said to have been presented by AFFiRiS’ Markus Mandler at the 2012 International Stockholm/Springfield Symposium on Advances in Alzheimer Therapy, although the conference abstract of his presentation[81] makes no mention of such: “Vaccinated mice retained twice as many dendrites as controls and about fifty percent more neurons. They outperformed controls in the Morris water maze test of spatial learning. The antibodies induced by PD01 somehow get across the blood brain barrier into the brain, said Mandler, because they turn up in neuronal lysosomes and in the perivascular space.”
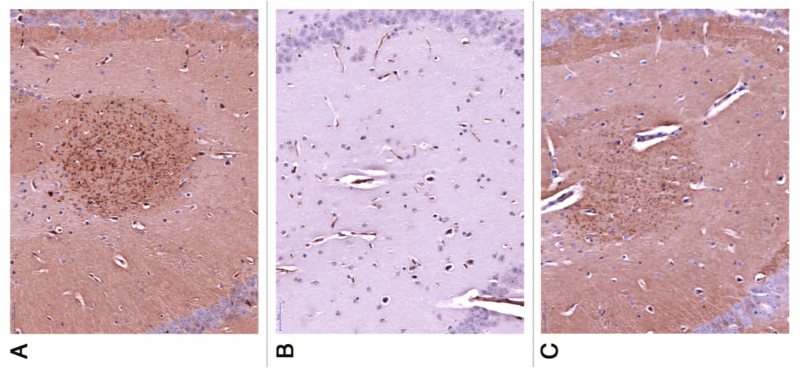
Figure 5. Vaccination with PD01 elicits antibodies recognizing human AS. Brain slides of human AS-transgenic mice exposed to serum from PD01-immunized WT mouse with (A) or without (B) a competing peptide (B), comparable to a commercially-available Ab specific to human AS. Reproduced from [47] under Creative Commons License from Landes Bioscience.
First-of-Class PD Vaccine Advances to Human Testing
Whatever its details, the accumulated evidence from preclinical testing was evidently sufficiently robust for European regulators to approve a Phase I clinical trial of PD01, and for MJFF to approve a $1.5M grant to help bring the AS-targeting immunotherapy into human testing. In June 2012, AFFiRiS announced the initiation of the trial.
The study center at the private Confraternität-Privatklinik Josefstadt hospital in Vienna recruited 32 volunteers with early-stage PD for testing of the safety and tolerability of a vaccine designated PD01A. Using a single-blind, parallel-group design, volunteers (12 in each group) have received (or soon will receive) subcutaneous injections of either a low (15 µg) or a high (75 µg) dose of the vaccine, once every four weeks for a year; up to eight additional subjects were also recruited as untreated controls. Patients in the trial are assessed quarterly on multiple safety, immunological, and potential clinical effects. Phase I trials are often carried out in healthy, young subjects to minimize patient risk, but as AFFiRiS scientists note,[49] a realistic test of safety for an active vaccine requires the presence of the target antigen in situ: eliciting an immune response to AS aggregates might be entirely harmless in young study volunteers who have negligible accumulations of the target antigen in their brains, but trigger life-threatening autoimmune attacks in subjects with the extensive cerebral AS aggregate deposition that characterizes clinical disease.
But while AFFiRiS chose the subjects of the first trial to be PD patients, it is notable that the trial design aims to recruit relatively young (45 to 65 y.o.) patients at genetic risk of the disease, and even younger (45-50 y.o.) patients in the absence of genetic risk but with a confirmed diagnosis of idiopathic PD — and the latter only after AFFiRiS approval. This is in line with one key lesson learned from the Aβ immunotherapy trials: the potential for clinical benefit is greatest, and the instantaneous risk of adverse events per unit time is lowest, when patients are relatively early in the disease course (or, better yet, during prodromal disease). This may be even more the case with PD than with AD: as noted previously, there is extensive AS pathology in the CNS (as well as the peripheral nervous system) in prodromal PD, well before SNc DA neuronal loss becomes extensive and major motor symptoms erupt (see Figure 1). At this point, the therapeutic target is present in the CNS, and its clearance may potentially prevent much of the loss of PD neurons that characterizes the mature disease. In those later stages, when neuronal loss is already extensive, a separate regenerative medicine approach (DA cell therapy) will be required to replace cells and circuitry that could have been rescued by early intervention, and the efficacy of AS immunotherapy may be masked by persistent motor symptoms even if nonmotor symptoms are substantially alleviated.
AFFiRiS has an additional reason to seek out study volunteers with determinative mutations for synucleinopathies that is more particular to PD than is the rationale for recruiting subjects with familial AD for the new Aβ immunotherapy trials. In the case of AD, several biomarkers have been established to allow for tracking of disease progression, target engagement, and disease modification, including CSF Aβ and tau and brain plaque imaged with either of two radioligands on positron emission tomography. By contrast, no such biomarkers yet exist for PD and other synucleinopathies.† Additionally, a vaccine targeting AS aggregates is most likely to be effective in prevention or arrest of the progression of the non-motor components of PD, which manifest earliest in the disease course, even if they become most troubling in its latest stages[14,20] — and yet validated scales for assessing nonmotor symptoms are just beginning to be constructed, proposed, and independently validated, and none have emerged as definitive.[61,62] By contrast, the Unified Parkinson’s Disease Rating Scale (UPDRS) is well-established for capturing the later-emerging motor symptoms of PD, which are thought to be principally driven by attrition of SNc dopaminergic neurons — a stage at which the optimal window for AS immunotherapy has been passed by.
The lack of robust biomarkers for progression of AS neuropathology or of well-established clinical testing scores for nonmotor symptoms will necessarily make the assessment of the disease-modifying activity of an AS-targeting vaccine more challenging than has already proven to be the case for Aβ immunotherapies. AFFiRiS researchers suggest that recruiting subjects with dominantly-inherited PD or other synucleinopathies might offer a means to overcome these challenges. First, genetic diagnosis and family history allow for early diagnosis many years before the clinical diagnosis of spontaneous PD cases can be made, by which point extensive neurodegeneration will already have occurred and the motor and nonmotor symptoms will be overlaid and difficult to disentangle clinically. Additionally, the clinical course of dominantly-inherited synucleinopathies is more homogeneous than in the more purely age-related disease, allowing for more confident evaluation of disease-modifying effects based on symptomatology alone.[50]
The original Phase I trial is evidently still ongoing, and anticipated to continue through December of 2013. AFFiRiS researchers have stated that they hope to gain “first insight into the immunological, biological and clinical activity in humans … encompass[ing] both motor and non-motor symptoms of the disease based on the fact that a disease-modifying drug is expected to act on all disease-affected domains.”[49] While an efficacy signal arising from so small and exploratory a trial would need to be treated as highly provisional, any sign of clinical benefit at this early stage would be welcome. Encouragingly, AFFiRiS announced a Phase Ib extension study in June of this year, to assess the long-term effects PD01A in treated patients in four quarterly visits for an additional year. This suggests, at minimum, that there were no adverse events sufficiently alarming as to lead to the discontinuation of the program in the original trial. The trial registration entry for the followup study specifies a wider range of motor and especially nonmotor symptoms for patient evaluation than were used in the original trial. One might alternately speculate that this casting of a wider net was a response to seeing little signal in the original trial’s outcome measures; a response to a positive signal more specific to nonmotor symptoms, as would be anticipated when relatively young subjects are given an effective AS-targeting rejuvenation biotechnology; or an expectation that a disease-modifying effect will by its nature become clearer over time, as the curves of clinical progression in the various study populations separate (see Figure 6).
For now, we eagerly await the results of this early, pioneering trial in rejuvenation biotechnology.
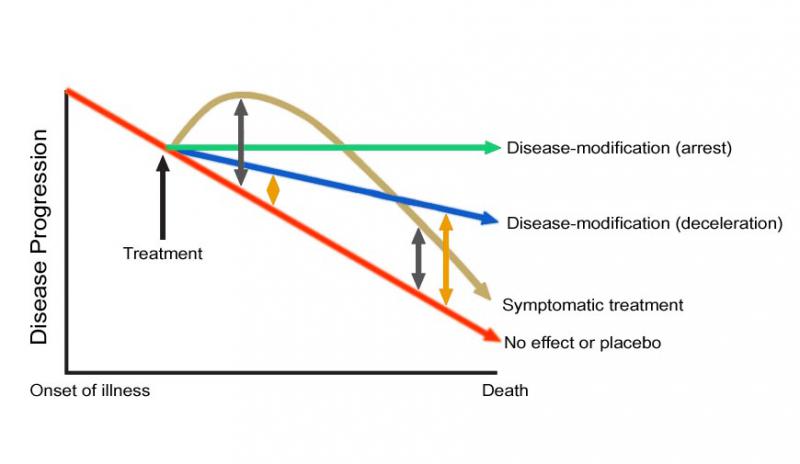
Figure 6. Effects of Disease-Modifying vs. Symptomatic Treatments in Progressive Diseases of Aging. The spread between symptomatically-treated and untreated subjects diminishes over time, as the structural basis of functions continue to degrade, while the spread increases over time for patients receiving disease-modifying therapies, as the accumulation of damage is decelerated or reversed.
Knowns and Unknowns on PD01(A)
As discussed above, developing neo-epitope vaccine antigens (AFFITOPEs) via the the AFFITOME technology has many conceptually attractive features. In principle, the AFFITOME system should enable the identification of novel antigens for rejuvenation immunotherapies that elicit a strong humoral immune response, while sidestepping a potentially disastrous Th1 autoimmune reaction. Evidently, both the preclinical safety and efficacy studies on PD01 and AFFiRiS’ three Aβ vaccines, and the Phase I clinical work to date, are consistent with this view, but no data have been formally published on any of this work, and attrition rates of new clinical entities are of course high in Phase II and Phase III.
The AFFITOME system should also be expected to deliver greatly-enhanced flexibility in designing and selecting candidate vaccine antigens, and in principle even to redirect the development pipeline as data emerge on a given vaccine in preclinical and even clinical development. AFFiRiS scientists have emphasized their intention to harness this flexibility to trial design, not only incorporating emerging scientific biological and clinical markers of disease progression into their ongoing clinical trial program,[50] but also an unusual willingness to test several such candidates at once. In a press release, AFFiRiS CSO Frank Mattner stated that “AFFITOME®-technology … delivers not only a single vaccine for the treatment of a certain disease but a whole pool of product candidates with excellent safety profiles and exactly fine tuned specificities. Therefore, we apply our strategy of “clinical maturation”, meaning that we investigate several vaccines against a certain disease in clinical testing to ensure that the best vaccine for humans will be developed.”
AFFiRiS have already demonstrated this diversification of risk and agility advancing new vaccines to trial in their AD program, within which three different Aβ-targeting AFFITOPEs have entered into human testing. In fact, the most recently-developed of the three candidates (MimoVax), which was designed in response to the strengthening momentum of scienctific evidence implicating N-terminally truncated and pyroglutamate-modified Aβ species in AD dementia,[63-69] has already caught up with and surpassed its two predecessors in clinical testing.
So the fact that the ongoing trials of AFFiRiS’ AS vaccine designate it as “PD01A” and not “PD01” as deemed in their scientific reports[49,50] and press releases would seem to indicate that it may already have been modified to some degree from their earliest human candidate, and that other AS-targeting vaccines may be under preclinical testing or waiting in the wings for human trials. (Compare the designations “AD01,” “AD02,” and “AD03” in their AD pipeline).
For example, it not entirely clear what species of AS PD01A targets, and one might speculate that there are several variants targeting different AS species (as previously was the case in their AD pipeline). AFFiRiS’ announcement of the PD01 Phase I trial says that “The vaccine called PD01A is directed against alpha-Synuclein,” which seems to indicate the protein in its physiological conformation. Consistent with this, one source[57] indicates that PD01 is “designed to trigger the body’s antibodies to attack and clear native alpha-synuclein proteins [my emphasis] stuck in the cell membrane or spreading between cells”,[57] and a second that it would induce the immune system to “generate antibodies against the vaccine that would then react to endogenous α-synuclein [again, emphasis mine].”[58] Such an approach might have motivated their elaboration of the fact that “α-syn knock-out mice are viable and show no overt neuronal phenotype, indicating that α-syn function can be replaced by other synucleins [and] β-syn shows the highest overlap of expression with α-syn” in one of their PD01-centered reviews,[47] on the hypothesis that redundant functions of β- and/or γ-synuclein might compensate for a vaccine-induced reduction of the steady-state level of physiological AS.
However, other sources indicate that PD01 directly targets AS aggregates. A review article on AS immunotherapy[56] cites AFFiRiS’ PD01 patent application (the full text of which is not public) as indicating that “PD01 is a mimotope that elicits a highly specific humoral immune response to clear an aberrant form of α-syn. PD01 targets the phosphorylated form of the protein, which activates microglia and induces neurotoxic proinflammatory responses.” MJFF has described the vaccine target in some blog posts as “alpha-synuclein clumps” but elsewhere says that “PD01 has been designed to target α-synuclein” (and again here). Neither AFFiRiS nor MJFF have responded to requests for comment on this apparent discrepancy.
Coalition-Building in Rejuvenation Research
As already noted above, the Michael J. Fox Foundation stepped in with funding at two key points in the development of PD01. In 2010, they supplied $475,000 to ensure that AFFiRiS would carry out further preclinical testing of the vaccine after AFFiRiS had already (prematurely?) announced that the PD01 preclinical program had been a success; in 2011, they would put down yet greater sums to help launch the Phase I human trial. In turn, their US$1.5 M commitment to support the PD01 Phase I trial drew in private investment from the venture capital firms Santo VC and MIG Funds, for a total private investment of €25M and an option to expand up to an additional €30M combined (Figure 7). As explained on the MJFF website, “In line with the Foundation’s mission to accelerate Parkinson’s disease research, we aim to de-risk the field, making it as attractive as possible for all researchers, particularly industry groups that will play a significant part in commercializing therapies. For the vast majority of the research projects we fund, we do not take a stake in intellectual property or revenue.“

Figure 7. Nonprofit Funding Played a Critical Role in Advancing PD01 to Clinical Testing. From the MJFF website.
The fact that it is the Michael J. Fox Foundation, in particular, that stepped in to support the development of PD01 is also a sign of the emerging recognition of the power of the “damage-repair” heuristic of rejuvenation biotechnology to prevent, arrest, and ultimately reverse the course of PD. Readers may recall that Michael J. Fox and his Foundation have played an important role in advancing embryonic stem cells and somatic cell nuclear transfer (SCNT/”therapeutic cloning”) as a key sources of cells for the medical use in PD and other crippling diseases of aging or inheritance. He and his foundation were prominent and valuable voices in the debate in the days leading up to and following the George W. Bush administration’s imposition of highly restrictive criteria for institutions receiving National Institutes of Health funding for embryonic stem cell research. They played similar roles in support of California’s Proposition 71, which established the California Institute of Regenerative Medicine as a source for billions of dollars in stable funding and responsible oversight for stem cell research in that state; in opposing a proposed United Nations ban on SCNT; and in support of political candidates favoring embryonic stem cell research against candidates who advocated for policies intended to hamper such research in Missouri and elsewhere. MJFF also made major early and ongoing investments in stem cell research when Federal funding was under severe restrictions. And while in recent years, it has reallocated its research budget consequent to the easing of funding restrictions and the threat of legislative bans on research, it still advocates for the new medical technology and says that “The Foundation will continue to monitor Parkinson’s disease-specific stem cell developments for opportunities where the Foundation can help in advancing this research.”
And as we have seen, MJFF’s subsequent investments have helped push forward the second key rejuvenation biotechnology for the prevention and cure of PD: immunotherapy targeting AS. SENS Research Foundation is committed to catalyzing the transformation of biomedical research to harness the power of regenerative medicine approaches to the entire sweep of the diseases and disabilities of aging. Key to this is the expanding recognition of the power of the “damage-repair” heuristic by major medical charities and patient advocacy groups like MJFF, who can help push regulators and incent the biomedical industry to develop the cures we all seek, and ultimately to achieve the extension of youthful health into a long life free of age-related disease.
Claiming — and Holding — the Yellow Jersey
This post has centered on PD01 as the first-in-class rejuvenation biotechnology targeting AS aggregates to advance into human trials. But while PD01 may be the first, there are competing vaccines using different approaches moving through the clinical pipeline. One of these competitors is Prothena’s PRX002, which appears to be (a humanized version of) one of the AS-targeting monoclonal antibodies tested in animal models by Masliah and Prothena’s Schenk([53], and see [57,70] and possibly [60]). The company’s webpage for PRX001 and their second quarter press release of August 16, 2013 indicate that it “Initiated pre-clinical IND enabling studies in second quarter of 2013” and that “IND filing and a Phase 1 trial in Parkinson’s disease patients are planned for 1H14 [first half of 2014],” a timeline consistent with a press report on Prothena including an interview with Schenk dating to late in 2012. A life sciences investment consulting firm indicates that “Preclinical data indicate that administration of PRX002 may slow or reduce neuropathology, possibly by preventing such transmission;”[58] the provenance of this information is unclear, as it is not mentioned in disclosures postdating that report.
Separately, BioArctic Neuroscience AB have recently reported early preclinical results with their lead candidate PD0805, which targets large AS oligomers[82] and evidently reduces the level of oligomeric AS species in the spinal cord of mice bearing human AS mutations[83]. And while the status of Biogen-Neurimmune’s AS-targeting antibody is unclear, it is clear that AFFiRiS will soon go from having the AS immunotherapy field to itself, to having to defend its head start against a growing field of competitors. Impressive as the ingenuity of the AFFITOME development platform may be, only clinical testing will reveal which of these many different candidates is the best approach for the safe and effective clearance of AS aggregates from the aging brain.
Building the Pyramid — And the Valley of Kings
The degenerative aging process is driven by the accumulation of multiple forms of cellular and molecular damage in the structures of our tissues, leading progressively over time to increasing disease, disability, and ultimately death. PD as a clinical entity emerges when the level of a specific subset of such lesions crosses of a clinical threshold. The key corollary of these facts is that when rejuvenation biotechnology matures to the point that this underlying damage is safely and effectively removed, repaired, replaced, or rendered harmless, then PD and the full spectrum of age-related disease and disability can be prevented, arrested in its course, and ultimately reversed.
AS vaccines will address one key driver of the PD phenotype: the accumulation of aggregated AS species in the brain and peripheral nervous system. But to fully arrest the progression of the disease, multiple rejuvenation biotechnologies will have to be brought to bear, each of them targeting specific cellular and molecular lesions involved in the constellation of pathology underlying PD. For instance, the evidence to date strongly suggests that these vaccines dispose of intracellular AS inclusions and/or “infective” extracellular AS aggregates by capturing them and tranporting them to the neuronal and/or microglial lysosome for degradation;[53,53,60] this suggests that these vaccines’ efficacy will be increased (or their ongoing effectiveness secured) by the therapeutic exploitation of microbial hydrolases to enhance the lysosome’s ability to degrade these and other recalcitrant cellular wastes.[74]
AS aggregates are strongly implicated in the progression of the nonmotor symptoms of PD and aging generally,[14-2072,73] but it is the age-related attrition of SNc DA neurons which is responsible for (most of) the classical motor symptoms of PD.[84,85] One driver of SNc DA neuronal loss is their accumulation to homoplasmy of mitochondria bearing large deletions in their DNA — a form of aging damage that occurs sporadically in cells of all long-lived tissues in the body, but which is especially prominent in the aging of SNc DA neurons. The deleterious effects of such mutations on oxidative tone and in contributing to the death of SNc DA neurons can be obviated through allotopic expression of the mitochondrially-encoded proteins and related strategies. Early intervention with AS vaccines to hold AS aggregate accumulation to levels beneath the “threshold of pathology” is also anticipated to reduce the rate of SNc DA neuronal loss, in addition to preventing or reversing the progression of the nonmotor symptoms of PD and aging generally.[14,72,73] Moreover, the rapid appearance of AS neuropathology in transplanted cells in the early trials of fetal cell therapy for PD[86] clearly suggests that clearance of these aggregates will be critical to securing maximum efficacy and durability of cell therapy.
However, neither AS immunotherapy nor the obviation of mitochondrial DNA deletions will abrogate outright the loss of SNc DA neurons with age (or, indeed, other neurons or long-lived cells in the body), and certainly they can do nothing to replace neurons that have already been lost to these or other aging processes when an aging person receives his or her first round of rejuvenation therapies. The development of safe and effective cell therapy to replace lost neurons and other cells is therefore critical to the prevention and cure of PD, and the loose pro-cure coalition that includes MJFF, SENS Research Foundation, and others must remain united to support research in the face of ongoing political opposition.
Happily, work in this area is proceeding apace. Moving forward from the promising but limited and mixed results of the first generation of cell therapies for PD,[71,76] recent years have seen the groundbreaking first transplantation of neurons derived by induced pluripotent stem cell (iPS) technology into the brains of nonhuman primates, and the parallel report that a cell population derived from human ESC can attenuate the Parkinsonian symptoms of cynomolgus monkeys treated with the mitochondrial toxin MPTP for at least 12 months post-transplant.[75] Researchers have also identified means to better differentiate such cells, and worked to understand and develop methods to eliminate other problems that plagued the early trials of fetal neuron transplants into PD patients.[76]
A key product of, and eventual contributor to, this translational research process is the recently-initiated TRANSEURO trial of fetal ventral mesencephalic cell grafts for people with PD. One hundred and twenty-one volunteers with early-stage PD were recruited at four European trial centers in the summer of 2012, and followed in an observational study to establish data on baseline status and disease course; of these, 20 were subsequently selected to receive cell grafts in an open-label Phase I trial. Learning from the experience of previous trials and subsequently-accumulated biological knowledge, the new trial has been designed to include “careful selection of patients (age, stage of disease, type of Parkinson’s disease), tissue preparation (number of cells grafted, dopaminergic vs serotonergic content of the graft), tissue placement (location, tract numbers), graft support (storage media, immunotherapy after grafting), and trial design (numbers of patients, follow-up time, endpoints).”[76] In fact, one of the goals of the Phase I trial is not just to implement a grafting protocol that incorporates these lessons learned, but to optimize the protocol through surgical and clinical experience with the volunteers. Promising results in the initial Phase I study will then trigger the initiation of a five-year, double-blind, sharm-surgery-controlled trial.[77]
Step by step, the rejuvenation biotechnologies needed to prevent and reverse the disabling neuropathology that drives PD are being developed, tested in rodents, and moved into clinical trials. The rejuvenation biotechnologies that emerge from these trials will initially be indicated for PD, but the lesions that these new therapies target are suffered by all aging people, driving the universal age-related loss of motor control, cognition, and autonomic dysfunction. Whether any individual suffers the obvious gait disturbances, tremors, and “masklike” loss of facial affect — these eventualities are in part a matter of individual variations in how rapidly the age-related lesions most specific to PD symptoms accumulate in our tissues, and in part a matter of the rate at which other aging damage accumulates in our bodies, overshadowing or pre-empting clinical PD with competiting forms of age-related morbidity and mortality.
An end to this, and to the misery of age-related ill health in all its forms, are what drives SENS Research Foundation’s work in research, education, outreach, transdisciplinary networking, and advocacy. We are encouraged by the progress being made in clearance of AS aggregates from the aging brain, moving us one step closer to the day when humanity will move free of the mummification of the years.
Acknowledgement
Thanks to Mattsson Web Solutions for graphics help.
Notes
*Other immunotherapy agents being tested in major AD preventive trials include gantenerumab and crenezumab.
†Despite this acknowledgement on the part of AFFiRiS researchers, the PD01 trial entry at ClinicalTrials.gov does list “Change in biological and radiological markers (e.g. CSF alpha synuclein levels)” as a measure of clinical activity of the vaccine. This is presumably being done in anticipation that this parameter will subsequently be validated as a marker of disease progression and/or target engagement, in which case baseline data and time sequences will prove invaluable. To their great credit, MJFF is very strategically funding what appear to be up to four approaches to develop imaging agents for LBs, and another to investigate CSF lysosomal hydrolases as potential CSF biomarkers. Robust biomarkers for CNS AS will greatly facilitate the development of rejuvenation biotechnologies targeting AS, and will be a major contribution to the field.
References
1: Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997 Jun 27;276(5321):2045-7. PubMed PMID: 9197268.
2: Krüger R, Kuhn W, Müller T, Woitalla D, Graeber M, Kösel S, Przuntek H, EpplenJT, Schöls L, Riess O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet. 1998 Feb;18(2):106-8. PubMed PMID: 9462735.
3: Zarranz JJ, Alegre J, Gómez-Esteban JC, Lezcano E,Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atarés B, Llorens V, Gomez Tortosa E, del Ser T, Muñoz DG, de Yebenes JG. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004 Feb;55(2):164-73. PubMed PMID: 14755719.
4: Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, Andrieux J, Hulihan M, Waucquier N, Defebvre L, Amouyel P, Farrer M, Destée A. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet. 2004 Sep 25-Oct 1;364(9440):1167-9. PubMed PMID: 15451224.
5: Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 2003 Oct 31;302(5646):841. PubMed PMID: 14593171.
6: Fuchs J, Nilsson C, Kachergus J, Munz M, Larsson EM, Schüle B, Langston JW, Middleton FA, Ross OA, Hulihan M, Gasser T, Farrer MJ. Phenotypic variation in a large Swedish pedigree due to SNCA duplication and triplication. Neurology. 2007 Mar 20;68(12):916-22. Epub 2007 Jan 24. PubMed PMID: 17251522.
7: Ross OA, Braithwaite AT, Skipper LM, Kachergus J, Hulihan MM, Middleton FA, Nishioka K, Fuchs J, Gasser T, Maraganore DM, Adler CH, Larvor L, Chartier-Harlin MC, Nilsson C, Langston JW, Gwinn K, Hattori N, Farrer MJ. Genomic investigation of alpha-synuclein multiplication and parkinsonism. Ann Neurol. 2008 Jun;63(6):743-50. doi: 10.1002/ana.21380. PubMed PMID: 18571778.
8: Ibáñez P, Lesage S, Janin S, Lohmann E, Durif F, Destée A, Bonnet AM, Brefel-Courbon C, Heath S, Zelenika D, Agid Y, Dürr A, Brice A; French Parkinson’s Disease Genetics Study Group. Alpha-synuclein gene rearrangements in dominantly inherited parkinsonism: frequency, phenotype, and mechanisms. Arch Neurol. 2009 Jan;66(1):102-8. doi: 10.1001/archneurol.2008.555. PubMed PMID: 19139307.
9: Miller DW, Hague SM, Clarimon J, Baptista M, Gwinn-Hardy K, Cookson MR, Singleton AB. Alpha-synuclein in blood and brain from familial Parkinson disease with SNCA locus triplication. Neurology. 2004 May 25;62(10):1835-8. PubMed PMID: 15159488.
10: International Parkinson Disease Genomics Consortium, Nalls MA, Plagnol V, Hernandez DG, Sharma M, Sheerin UM, Saad M, Simón-Sánchez J,Schulte C, Lesage S, Sveinbjörnsdóttir S, Stefánsson K, Martinez M, Hardy J, Heutink P, Brice A, Gasser T, Singleton AB, Wood NW. Imputation of sequence variants for identification of genetic risks for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet. 2011 Feb 19;377(9766):641-9. doi: 10.1016/S0140-6736(10)62345-8. Epub 2011 Feb 1. PubMed PMID: 21292315; PubMed Central PMCID: PMC3696507.
10a: Sharma M, Ioannidis JP, Aasly JO, Annesi G, Brice A, Van Broeckhoven C, Bertram L, Bozi M, Crosiers D, Clarke C, Facheris M, Farrer M, Garraux G, Gispert S, Auburger G, Vilariño-Güell C, Hadjigeorgiou GM, Hicks AA, Hattori N, Jeon B, Lesage S, Lill CM, Lin JJ, Lynch T, Lichtner P, Lang AE, Mok V, Jasinska-Myga B, Mellick GD, Morrison KE, Opala G, Pramstaller PP, Pichler I, Park SS, Quattrone A, Rogaeva E, Ross OA, Stefanis L, Stockton JD, Satake W, Silburn PA, Theuns J, Tan EK, Toda T, Tomiyama H, Uitti RJ, Wirdefeldt K, Wszolek Z, Xiromerisiou G, Yueh KC, Zhao Y, Gasser T, Maraganore D, Krüger R; GEO-PD Consortium. Large-scale replication and heterogeneity in Parkinson disease genetic loci. Neurology. 2012 Aug 14;79(7):659-67. doi: 10.1212/WNL.0b013e318264e353. Epub 2012 Jul 11. PubMed PMID: 22786590; PubMed Central PMCID: PMC3414661.
11: Maraganore DM, de Andrade M, Elbaz A, Farrer MJ, Ioannidis JP, Krüger R, Rocca WA, Schneider NK, Lesnick TG, Lincoln SJ, Hulihan MM, Aasly JO, Ashizawa T, Chartier-Harlin MC, Checkoway H, Ferrarese C, Hadjigeorgiou G, Hattori N, Kawakami H, Lambert JC, Lynch T, Mellick GD, Papapetropoulos S, Parsian A, Quattrone A, Riess O, Tan EK, Van Broeckhoven C; Genetic Epidemiology of Parkinson’s Disease (GEO-PD) Consortium. Collaborative analysis of alpha-synuclein gene promoter variability and Parkinson disease. JAMA. 2006 Aug 9;296(6):661-70. PubMed PMID: 16896109.
12: Mueller JC, Fuchs J, Hofer A, Zimprich A, Lichtner P, Illig T, Berg D, Wüllner U, Meitinger T, Gasser T. Multiple regions of alpha-synuclein are associated with Parkinson’s disease. Ann Neurol. 2005 Apr;57(4):535-41. PubMed PMID: 15786467.
13: Rhinn H, Qiang L, Yamashita T, Rhee D, Zolin A, Vanti W, Abeliovich A. Alternative α-synuclein transcript usage as a convergent mechanism in Parkinson’s disease pathology. Nat Commun. 2012;3:1084. doi: 10.1038/ncomms2032. PubMed PMID: 23011138; PubMed Central PMCID: PMC3660047.
14: Lim SY, Fox SH, Lang AE. Overview of the extranigral aspects of Parkinson disease. Arch Neurol. 2009 Feb;66(2):167-72. doi: 10.1001/archneurol.2008.561. Review. PubMed PMID: 19204152.
15: Markesbery WR, Jicha GA, Liu H, Schmitt FA. Lewy body pathology in normal elderly subjects. J Neuropathol Exp Neurol. 2009 Jul;68(7):816-22. doi: 10.1097/NEN.0b013e3181ac10a7. PubMed PMID: 19535990; PubMed Central PMCID: PMC2704264.
16: Ferrer I, Martinez A, Blanco R, Dalfó E, Carmona M. Neuropathology of sporadic Parkinson disease before the appearance of parkinsonism: preclinical Parkinson disease. J Neural Transm. 2011 May;118(5):821-39. doi: 10.1007/s00702-010-0482-8. Epub 2010 Sep 23. Review. PubMed PMID: 20862500.
17: Del Tredici K, Hawkes CH, Ghebremedhin E, Braak H. Lewy pathology in the submandibular gland of individuals with incidental Lewy body disease and sporadic Parkinson’s disease. Acta Neuropathol. 2010 Jun;119(6):703-13. doi: 10.1007/s00401-010-0665-2. Epub 2010 Mar 14. PubMed PMID: 20229352.
18: Savica R, Carlin JM, Grossardt BR, Bower JH, Ahlskog JE, Maraganore DM, Bharucha AE, Rocca WA. Medical records documentation of constipation preceding Parkinson disease: A case-control study. Neurology. 2009 Nov 24;73(21):1752-8. doi: 10.1212/WNL.0b013e3181c34af5. PubMed PMID: 19933976; PubMed Central PMCID: PMC2788809.
19: Abbott RD, Ross GW, Petrovitch H, Tanner CM, Davis DG, Masaki KH, Launer LJ, Curb JD, White LR. Bowel movement frequency in late-life and incidental Lewy bodies. Mov Disord. 2007 Aug 15;22(11):1581-6. PubMed PMID: 17523195.
20: Stern MB, Lang A, Poewe W. Toward a redefinition of Parkinson’s disease. Mov Disord. 2012 Jan;27(1):54-60. doi: 10.1002/mds.24051. PubMed PMID: 22252891.
21: Crabtree DM, Zhang J. Genetically engineered mouse models of Parkinson’s disease. Brain Res Bull. 2012 May 1;88(1):13-32. doi: 10.1016/j.brainresbull.2011.07.019. Epub 2011 Aug 3. Review. PubMed PMID: 21839151; PubMed Central PMCID: PMC3244549.
22: Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara Y, Sisk A, Mucke L. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science. 2000 Feb 18;287(5456):1265-9. PubMed PMID: 10678833.
23: Marques O, Outeiro TF. Alpha-synuclein: from secretion to dysfunction and death. Cell Death Dis. 2012 Jul 19;3:e350. doi: 10.1038/cddis.2012.94. Review. PubMed PMID: 22825468; PubMed Central PMCID: PMC340659.
24: Lotharius J, Brundin P. Pathogenesis of Parkinson’s disease: dopamine, vesicles and alpha-synuclein. Nat Rev Neurosci. 2002 Dec;3(12):932-42. Review. PubMed PMID: 12461550.
25: Doty RL. Olfactory dysfunction in Parkinson disease. Nat Rev Neurol. 2012 May 15;8(6):329-39. doi: 10.1038/nrneurol.2012.80. Review. PubMed PMID: 22584158.
26: Halliday G, Lees A, Stern M. Milestones in Parkinson’s disease–clinical and pathologic features. Mov Disord. 2011 May;26(6):1015-21. doi: 10.1002/mds.23669. Review. PubMed PMID: 21626546.
27: Chu Y, Kordower JH. Age-associated increases of alpha-synuclein in monkeys and humans are associated with nigrostriatal dopamine depletion: Is this the target for Parkinson’s disease? Neurobiol Dis. 2007 Jan;25(1):134-49. Epub 2006 Oct 18. PubMed PMID: 17055279.
28: Markopoulou K, Ahlskog JE, Anderson KJ, Armasu SM, Biernacka JM, Chung SJ, Cunningham JM, Frigerio R, Maraganore DM. Genetic evidence for a dual and opposing effect of alpha-synuclein expression in preclinical versus clinical Parkinson’s disease. Neurology. 2013 Feb 12; 80(Meeting Abstracts 1): IN2-1.003.
29: Ritz B, Rhodes SL, Bordelon Y, Bronstein J. α-Synuclein genetic variants predict faster motor symptom progression in idiopathic Parkinson disease. PLoS One. 2012;7(5):e36199. doi: 10.1371/journal.pone.0036199. Epub 2012 May 15. PubMed PMID: 22615757; PubMed Central PMCID: PMC3352914.
30: Morris M, Koyama A, Masliah E, Mucke L. Tau reduction does not prevent motor deficits in two mouse models of Parkinson’s disease. PLoS One. 2011;6(12):e29257. doi: 10.1371/journal.pone.0029257. Epub 2011 Dec 19. PubMed PMID: 22206005; PubMed Central PMCID: PMC3242771.
31: Roberson ED, Halabisky B, Yoo JW, Yao J, Chin J, Yan F, Wu T, Hamto P, Devidze N, Yu GQ, Palop JJ, Noebels JL, Mucke L. Amyloid-β/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J Neurosci. 2011 Jan 12;31(2):700-11. doi: 10.1523/JNEUROSCI.4152-10.2011. PubMed PMID: 21228179; PubMed Central PMCID: PMC3325794.
32: Andrews-Zwilling Y, Bien-Ly N, Xu Q, Li G, Bernardo A, Yoon SY, Zwilling D, Yan TX, Chen L, Huang Y. Apolipoprotein E4 causes age- and Tau-dependent impairment of GABAergic interneurons, leading to learning and memory deficits in mice. J Neurosci. 2010 Oct 13;30(41):13707-17. doi: 10.1523/JNEUROSCI.4040-10.2010. PubMed PMID: 20943911; PubMed Central PMCID: PMC2988475.
33: Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wölfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Götz J. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell. 2010 Aug 6;142(3):387-97. doi: 10.1016/j.cell.2010.06.036. Epub 2010 Jul 22. PubMed PMID: 20655099.
34: Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science. 2007 May 4;316(5825):750-4. PubMed PMID: 17478722.
35: Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005 Jul 15;309(5733):476-81. PubMed PMID: 16020737; PubMed Central PMCID: PMC1574647.
36: Duka T, Duka V, Joyce JN, Sidhu A. Alpha-Synuclein contributes to GSK-3beta-catalyzed Tau phosphorylation in Parkinson’s disease models. FASEB J. 2009 Sep;23(9):2820-30. doi: 10.1096/fj.08-120410. Epub 2009 Apr 15. PubMed PMID: 19369384; PubMed Central PMCID: PMC2796901.
37: Duka T, Rusnak M, Drolet RE, Duka V, Wersinger C, Goudreau JL, Sidhu A. Alpha-synuclein induces hyperphosphorylation of Tau in the MPTP model of parkinsonism. FASEB J. 2006 Nov;20(13):2302-12. PubMed PMID: 17077307.
38: Lin CH, Tsai PI, Wu RM, Chien CT. LRRK2 G2019S mutation induces dendrite degeneration through mislocalization and phosphorylation of tau by recruiting autoactivated GSK3ß. J Neurosci. 2010 Sep 29;30(39):13138-49. doi: 10.1523/JNEUROSCI.1737-10.2010. PubMed PMID: 20881132.
39: Lei P, Ayton S, Finkelstein DI, Spoerri L, Ciccotosto GD, Wright DK, Wong BX, Adlard PA, Cherny RA, Lam LQ, Roberts BR, Volitakis I, Egan GF, McLean CA, Cappai R, Duce JA, Bush AI. Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat Med. 2012 Jan 29;18(2):291-5. doi: 10.1038/nm.2613. PubMed PMID: 22286308.
40: Lobello K, Ryan JM, Liu E, Rippon G, Black R. Targeting Beta amyloid: a clinical review of immunotherapeutic approaches in Alzheimer’s disease. Int J Alzheimers Dis. 2012;2012:628070. doi: 10.1155/2012/628070. Epub 2012 Jan 15. PubMed PMID: 22292124; PubMed Central PMCID: PMC3265072.
41: Krishnamurthy PK, Deng Y, Sigurdsson EM. Mechanistic Studies of Antibody-Mediated Clearance of Tau Aggregates Using an ex vivo Brain Slice Model. Front Psychiatry. 2011 Oct 21;2:59. doi: 10.3389/fpsyt.2011.00059. PubMed PMID: 22025915; PubMed Central PMCID: PMC3198029.
42: de Calignon A, Polydoro M, Suárez-Calvet M, William C, Adamowicz DH, Kopeikina KJ, Pitstick R, Sahara N, Ashe KH, Carlson GA, Spires-Jones TL, Hyman BT. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron. 2012 Feb 23;73(4):685-97. doi: 10.1016/j.neuron.2011.11.033. Erratum in: Neuron. 2012 Oct 18;76(2):461. PubMed PMID: 22365544; PubMed Central PMCID: PMC3292759.
43: Kfoury N, Holmes BB, Jiang H, Holtzman DM, Diamond MI. Trans-cellular propagation of Tau aggregation by fibrillar species. J Biol Chem. 2012 Jun 1;287(23):19440-51. doi: 10.1074/jbc.M112.346072. Epub 2012 Mar 29. PubMed PMID: 22461630; PubMed Central PMCID: PMC3365982.
44: Monsonego A, Zota V, Karni A, Krieger JI, Bar-Or A, Bitan G, Budson AE, Sperling R, Selkoe DJ, Weiner HL. Increased T cell reactivity to amyloid beta protein in older humans and patients with Alzheimer disease. J Clin Invest. 2003 Aug;112(3):415-22. PubMed PMID: 12897209; PubMed Central PMCID: PMC166296.
45: Hashimoto M, Rockenstein E, Mante M, Mallory M, Masliah E. beta-Synuclein inhibits alpha-synuclein aggregation: a possible role as an anti-parkinsonian factor. Neuron. 2001 Oct 25;32(2):213-23. PubMed PMID: 11683992.
46: Tsigelny IF, Bar-On P, Sharikov Y, Crews L, Hashimoto M, Miller MA, Keller SH, Platoshyn O, Yuan JX, Masliah E. Dynamics of alpha-synuclein aggregation and inhibition of pore-like oligomer development by beta-synuclein. FEBS J. 2007 Apr;274(7):1862-77. PubMed PMID: 17381514.
47: Schneeberger A, Mandler M, Mattner F, Schmidt W. AFFITOME® technology in neurodegenerative diseases: the doubling advantage. Hum Vaccin. 2010 Nov;6(11):948-52. Epub 2010 Nov 1. Review. PubMed PMID: 20980801.
48: Schneeberger A, Mandler M, Otawa O, Zauner W, Mattner F, Schmidt W. Development of AFFITOPE vaccines for Alzheimer’s disease (AD)–from concept to clinical testing. J Nutr Health Aging. 2009 Mar;13(3):264-7. Review. PubMed PMID: 19262965.
49: Schneeberger A, Mandler M, Mattner F, Schmidt W. Vaccination for Parkinson’s disease. Parkinsonism Relat Disord. 2012 Jan;18 Suppl 1:S11-3. doi: 10.1016/S1353-8020(11)70006-2. Review. PubMed PMID: 22166404.
50: Schneeberger A, Kutzelnigg A, Mandler M. Parkinson’s disease vaccine: clinical trial challenges when striving for disease modification. Clin Invest. 2013 Jan;3(1):1-4.
51: Lemere CA, Masliah E. Can Alzheimer disease be prevented by amyloid-beta immunotherapy? Nat Rev Neurol. 2010 Feb;6(2):108-19. PubMed PMID: 20140000; PubMed Central PMCID: PMC2864089.
52: Masliah E, Rockenstein E, Adame A, Alford M, Crews L, Hashimoto M, Seubert P, Lee M, Goldstein J, Chilcote T, Games D, Schenk D. Effects of alpha-synuclein immunization in a mouse model of Parkinson’s disease. Neuron. 2005 Jun 16;46(6):857-68. PubMed PMID: 15953415.
53: Masliah E, Rockenstein E, Mante M, Crews L, Spencer B, Adame A, Patrick C, Trejo M, Ubhi K, Rohn TT, Mueller-Steiner S, Seubert P, Barbour R, McConlogue L, Buttini M, Games D, Schenk D. Passive immunization reduces behavioral and neuropathological deficits in an alpha-synuclein transgenic model of Lewy body disease. PLoS One. 2011 Apr 29;6(4):e19338. doi: 10.1371/journal.pone.0019338. PubMed PMID: 21559417; PubMed Central PMCID: PMC3084838.
54: Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008 May 30;133(5):775-87. doi: 10.1016/j.cell.2008.05.009. Review. PubMed PMID: 18510923.
55: Tomljenovic L, Shaw CA. Aluminum vaccine adjuvants: are they safe? Curr Med Chem. 2011;18(17):2630-7. PubMed PMID: 21568886.
56: Hutter-Saunders JA, Mosley RL, Gendelman HE. Pathways towards an effective immunotherapy for Parkinson’s disease. Expert Rev Neurother. 2011 Dec;11(12):1703-15. doi: 10.1586/ern.11.163. Review. PubMed PMID: 22091596; PubMed Central PMCID: PMC3263336.
57: Dolgin E. First therapy targeting Parkinson’s proteins enters clinical trials. Nat Med. 2012 Jul 6;18(7):992-3. doi: 10.1038/nm0712-992b. PubMed PMID: 22772533.
58: McDonald AI, Isaacson J, Washington C. LifeSci Advisors. LifeSci Investment Abstract: Prothena Corporation plc. Initiation of Coverage. Prothena. 2013 May 15.
59: Paul S, Planque S, Nishiyama Y. Immunological origin and functional properties of catalytic autoantibodies to amyloid beta peptide. J Clin Immunol. 2010 May;30 Suppl 1:S43-9. PubMed PMID: 20454852.
60: Bae EJ, Lee HJ, Rockenstein E, Ho DH, Park EB, Yang NY, Desplats P, Masliah E, Lee SJ. Antibody-aided clearance of extracellular α-synuclein prevents cell-to-cell aggregate transmission. J Neurosci. 2012 Sep 26;32(39):13454-69. PubMed PMID: 23015436; PubMed Central PMCID: PMC3752153.
61: Martinez-Martin P, Chaudhuri KR, Rojo-Abuin JM, Rodriguez-Blazquez C, Alvarez-Sanchez M, Arakaki T, Bergareche-Yarza A, Chade A, Garretto N, Gershanik O, Kurtis MM, Martinez-Castrillo JC, Mendoza-Rodriguez A, Moore HP, Rodriguez-Violante M, Singer C, Tilley BC, Huang J, Stebbins GT, Goetz CG. Assessing the non-motor symptoms of Parkinson’s disease: MDS-UPDRS and NMS Scale. Eur J Neurol. 2013 Apr 22. doi: 10.1111/ene.12165. [Epub ahead of print] PubMed PMID: 23607783.
62: Buck PO, Trautman H, Clark J. Scales for assessing nonmotor symptom severity changes in Parkinson’s disease patients with symptom fluctuations. Int J Neurosci. 2010 Aug;120(8):523-30. doi: 10.3109/00207454.2010.489725. Review. PubMed PMID: 20615055.
62: Benner EJ, Banerjee R, Reynolds AD, Sherman S, Pisarev VM, Tsiperson V, Nemachek C, Ciborowski P, Przedborski S, Mosley RL, Gendelman HE. Nitrated alpha-synuclein immunity accelerates degeneration of nigral dopaminergic neurons. PLoS One. 2008 Jan 2;3(1):e1376. doi: 10.1371/journal.pone.0001376. PubMed PMID: 18167537; PubMed Central PMCID: PMC2147051.
63: Schilling S, Zeitschel U, Hoffmann T, Heiser U, Francke M, Kehlen A, Holzer M, Hutter-Paier B, Prokesch M, Windisch M, Jagla W, Schlenzig D, Lindner C, Rudolph T, Reuter G, Cynis H, Montag D, Demuth HU, Rossner S. Glutaminyl cyclase inhibition attenuates pyroglutamate Abeta and Alzheimer’s disease-like pathology. Nat Med. 2008 Oct;14(10):1106-11. doi: 10.1038/nm.1872. Epub 2008 Sep 28. PubMed PMID: 18836460.
64: Siemers ER, Friedrich S, Dean RA, Sethuraman G, DeMattos RM, Jennings D, Tamagnan G, Marek K, Seibyl J. Alterations Of Novel Abeta Biomarkers After Administration Of A Monoclonal Antibody Specific For Soluble Abeta. Alzheimers Dement. 2008 Jul;4(4 Suppl): T774 (Abs P4-346). doi:10.1016/j.jalz.2008.05.2416
65:Piccini A, Russo C, Gliozzi A, Relini A, Vitali A, Borghi R, Giliberto L, Armirotti A, D’Arrigo C, Bachi A, Cattaneo A, Canale C, Torrassa S, Saido TC, Markesbery W, Gambetti P, Tabaton M. beta-amyloid is different in normal aging and in Alzheimer disease. J Biol Chem. 2005 Oct 7;280(40):34186-92. Epub 2005 Aug 15. PubMed PMID: 16103127.
66: Vanderstichele H, De Meyer G, Andreasen N, Kostanjevecki V, Wallin A, Olsson A, Blennow K, Vanmechelen E. Amino-truncated beta-amyloid42 peptides in cerebrospinal fluid and prediction of progression of mild cognitive impairment. Clin Chem. 2005 Sep;51(9):1650-60. Epub 2005 Jul 14. PubMed PMID: 16020497.
67: Harigaya Y, Saido TC, Eckman CB, Prada CM, Shoji M, Younkin SG. Amyloid beta protein starting pyroglutamate at position 3 is a major component of the amyloid deposits in the Alzheimer’s disease brain. Biochem Biophys Res Commun. 2000 Sep 24;276(2):422-7. PubMed PMID: 11027491.
68: Saido TC, Iwatsubo T, Mann DM, Shimada H, Ihara Y, Kawashima S. Dominant and differential deposition of distinct beta-amyloid peptide species, A beta N3(pE), in senile plaques. Neuron. 1995 Feb;14(2):457-66. PubMed PMID: 7857653.
69: Roher AE, Lowenson JD, Clarke S, Wolkow C, Wang R, Cotter RJ, Reardon IM, Zürcher-Neely HA, Heinrikson RL, Ball MJ, et al. Structural alterations in the peptide backbone of beta-amyloid core protein may account for its deposition and stability in Alzheimer’s disease. J Biol Chem. 1993 Feb 15;268(5):3072-83. PubMed PMID: 8428986.
70: Games D, Seubert P, Rockenstein E, Patrick C, Trejo M, Ubhi K, Ettle B, Ghassemiam M, Barbour R, Schenk D, Nuber S, Masliah E. Axonopathy in an α-synuclein transgenic model of Lewy body disease is associated with extensive accumulation of C-terminal-truncated α-synuclein. Am J Pathol. 2013 Mar;182(3):940-53. doi: 10.1016/j.ajpath.2012.11.018. Epub 2013 Jan 9. PubMed PMID: 23313024; PubMed Central PMCID: PMC3589076.
71: Braak H, Del Tredici K. Assessing fetal nerve cell grafts in Parkinson’s disease. Nat Med. 2008 May;14(5):483-5. PubMed PMID: 18463652.
72: Levy G. The relationship of Parkinson disease with aging. Arch Neurol. 2007 Sep;64(9):1242-6. Review. PubMed PMID: 17846263.
73: Lang AE, Obeso JA. Challenges in Parkinson’s disease: restoration of the nigrostriatal dopamine system is not enough. Lancet Neurol. 2004 May;3(5):309-16. Review. PubMed PMID: 15099546.
74: Mathieu JM, Schloendorn J, Rittmann BE, Alvarez PJ. Medical bioremediation of age-related diseases. Microb Cell Fact. 2009 Apr 9;8:21. PubMed PMID: 19358742; PubMed Central PMCID: PMC2674406.
75: Doi D, Morizane A, Kikuchi T, Onoe H, Hayashi T, Kawasaki T, Motono M, Sasai Y, Saiki H, Gomi M, Yoshikawa T, Hayashi H, Shinoyama M, Refaat MM, Suemori H, Miyamoto S, Takahashi J. Prolonged maturation culture favors a reduction in the tumorigenicity and the dopaminergic function of human ESC-derived neural cells in a primate model of Parkinson’s disease. Stem Cells. 2012 May;30(5):935-45. doi: 10.1002/stem.1060. PubMed PMID: 22328536.
76: Barker RA, Barrett J, Mason SL, Björklund A. Fetal dopaminergic transplantation trials and the future of neural grafting in Parkinson’s disease. Lancet Neurol. 2013 Jan;12(1):84-91. doi: 10.1016/S1474-4422(12)70295-8. Review. PubMed PMID: 23237903.
77: Lindvall O, Björklund A. Cell therapeutics in Parkinson’s disease. Neurotherapeutics. 2011 Oct;8(4):539-48. doi: 10.1007/s13311-011-0069-6. Review. PubMed PMID: 21901584; PubMed Central PMCID: PMC3250287.
78: de Grey AD, Ames BN, Andersen JK, Bartke A, Campisi J, Heward CB, McCarter RJM, Stock G. Time to talk SENS: critiquing the immutability of human aging. Ann N Y Acad Sci 2002;959:452-462. PMID: 11976218 [PubMed – indexed for MEDLINE]
79: de Grey AD. An engineer’s approach to the development of real anti-aging medicine. Sci Aging Knowledge Environ. 2003 Jan 8;2003(1):VP1. PMID: 12844502 [PubMed – indexed for MEDLINE]
80: Rae MJ. SENS Foundation: Accelerating Progress toward Biomedical Rejuvenation. In Fahy GM, West M, Coles LS, Harris SB (eds). The Future of Aging. 2010; Springer, New York, NY. pp. 367–438. doi:10.1007/978-90-481-3999-6_12. ISBN 978-90-481-3998-9.
81: Mandler M, Schneeberger A, Schmidt W, Mattner F. AFFITOPE®-based vaccines: a novel and safe immunotherapeutic strategy for neurodegenerative diseases. Presentation at the 12th International Stockholm/Springfield Symposium on Advances in Alzheimer Therapy. 2012 May 12;55.
82: Fagerqvist T, Lindström V, Nordström E, Lord A, Tucker SM, Su X, Sahlin C, Kasrayan A, Andersson J, Welander H, Näsström T, Holmquist M, Schell H, Kahle PJ, Kalimo H, Möller C, Gellerfors P, Lannfelt L, Bergström J, Ingelsson M. Monoclonal antibodies selective for α-synuclein oligomers/protofibrils recognize brain pathology in Lewy body disorders and α-synuclein transgenic mice with the disease-causing A30P mutation. J Neurochem. 2013 Jul;126(1):131-44. doi: 10.1111/jnc.12175. Epub 2013 Feb 27. PubMed PMID: 23363402.
83: Fagerqvist T. Studies of α-synuclein Oligomers — with Relevance to Lewy Body Disorders. Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Medicine 925. 2013; Acta Universitatis Upsaliensis, Uppsala. ISSN 1651-6206 ISBN 978-91-554-8720-1
84: Greffard S, Verny M, Bonnet AM, Beinis JY, Gallinari C, Meaume S, Piette F, Hauw JJ, Duyckaerts C. Motor score of the Unified Parkinson Disease Rating Scale as a good predictor of Lewy body-associated neuronal loss in the substantia nigra. Arch Neurol. 2006 Apr;63(4):584-8. PubMed PMID: 16606773.
85: Ma SY, Röyttä M, Rinne JO, Collan Y, Rinne UK. Correlation between neuromorphometry in the substantia nigra and clinical features in Parkinson’s disease using disector counts. J Neurol Sci. 1997 Oct 3;151(1):83-7. PubMed PMID: 9335015.
86: Braak H, Del Tredici K. Assessing fetal nerve cell grafts in Parkinson’s disease. Nat Med. 2008 May;14(5):483-5. PubMed PMID: 18463652.