
Short summary: Aberrant tau inside neurons is a key driver of Alzheimer’s disease, but nearly all the therapies in development to target it can only capture the small amount that floats outside of them. A new animal study reports impressive results in clearing aberrant tau inside neurons and rejuvenating cognitive function, opening up an important new front in damage-repair strategies for maintaining the aging brain.
An apocryphal story has it that when a reporter asked Willie Sutton why he robbed banks, the dashing heist artist replied, “Because that’s where the money is.” The pharmaceutical industry has hitherto studiously neglected Sutton’s Rule in their efforts to target aberrant tau.
We now have effective AmyloSENS therapies that directly remove soluble beta-amyloid from the brain and thereby slow down the terrible downward trajectory of neurodegenerative aging of the Alzheimer’s type (AD). The critical next target for delaying, preventing, and eventually reversing AD is abnormal forms of the protein tau. While beta-amyloid is the root driver of this variant of brain aging, that damaged protein wreaks much of its havoc by driving aberrant tau into the neocortex, the seat of our memories and our sense of ourselves.
Even before people have a clinical diagnosis of AD, people with so-called “mild” cognitive impairment (MCI) are already burdened by substantial amounts of aberrant tau in the regions that predispose a person to outright dementia. (Despite the name, MCI is a substantial and disabling level of cognitive dysfunction). And while the clinical trials for donanemab (Kisunla®) and lecanemab (Leqembi®) showed that clearing beta-amyloid out of the brain can prevent even more of this downstream damage from occurring, it can’t remove the existing tau damage laid down earlier in the disease process. For that, we need direct tau-clearing rejuvenation biotechnologies.
In addition to the benefits of tau clearance for its own sake, data from the donanemab and lecanemab clinical trials revealed that the lower a person’s burden of aberrant tau, the better these rejuvenation biotechnologies work. The clear implication is that combining a beta-amyloid-removal therapy with a LysoSENS therapy that purges the brain of aberrant tau would substantially increase the effectiveness of amyloid clearance, especially in people with higher baseline levels of tau damage.
The good news is that multiple biotech companies, from new startups to established players with Big Pharma partners, are advancing their own candidate tau-targeting therapies through the clinical pipeline. And while companies abandoned a number of these would-be therapies early on because they interfered with the physiological function of the healthy, non-aggregated tau protein, many such candidates are now in early- to mid-stage human clinical trials.
But as we discussed in a previous post, most of these tau-targeting therapies have a critical limitation: they only clear tau aggregates located outside of neurons, either in the gaps between neurons where they communicate with others in their neighborhood or in the bulk fluid that bathes the brain. Such therapies should do some good, most notably by slowing down the rate at which “seeds” of aberrant tau “infect” neurons with which they are in contact. But they would do nothing to clear existing aberrant tau inside of neurons, which is where those seeds are formed, where most of the aberrant tau is located, and where it inflicts most of its harm. Unsurprisingly, such tau immunotherapies become ineffective once advanced tau pathology takes hold in the brain.
In a recent post, we covered a very preliminary study by Australian scientists seeking to use an antibody mRNA to target tau inside neurons. It was a promising early result, and foreshadowed not only the ability to hit tau aggregates where they live, but to use mRNA to deliver other rejuvenation biotechnologies against damaged molecules inside neurons. But there were some important limitations to the technology they had developed: they lacked a strategy for getting their therapeutic mRNA into the brain, they only had results in cell culture, and their antibody was not specific to aberrant tau, but instead hit even healthy tau indiscriminately.
Now the productive group led by Rakez Kayed at the University of Texas Medical Branch has developed a candidate that seems to solve all of these problems, creating hope that aberrant tau could soon be the next form of molecular aging damage to fall to rejuvenation biotechnology.
Their potential LysoSENS therapy, called toxic tau conformation–specific monoclonal antibody-2 (TTCM2), has two important design elements that address the main limitations of the tau therapies that are currently in clinical trials. First, they let biology design their antibody for them, instead of trying to craft one based on their theoretical expectations. Too often, researchers have designed therapeutic antibodies to target specific segments of the tau protein that are either held in common with healthy tau (and thus capture the functional protein along with aberrant species) or that turn out to be so buried into the warped structure of tau aggregates that the antibody can’t reach it.
Instead, Kayed and colleagues cooked up some soluble tau oligomers, which are likely the main culprits in driving cognitive dysfunction and toxicity to neurons, and turned them into a “vaccine” by injecting them into mice. This caused the mice’s immune system to create antibodies targeting these oligomers. The team then used these antibodies as the core of their therapy, knowing that they would bind to the kind of tau they wanted to clear out.
As an initial test of whether these antibodies would bind to real aberrant tau in the brain and not just to synthetic tau oligomers in the plasma of mice, the team then applied them to brain sections taken from people who had died with AD or with other neurodegenerative aging diseases that are driven by aberrant tau. Sure enough, TTCM2 antibodies zeroed in on locations where aggregated tau was present in all three of these brain aging disorders, but conversely found nothing to latch onto in the brains of nondemented control subjects.
The fact that TTCM2 binds to tau monomers at all initially seems less than ideal, as tau monomers are typically the healthy, normal tau protein that the brain needs to build and maintain the cellular monorail for shuttling proteins and organelles around, among other functions of tau. Indeed, the loss of normal tau monomers when they get stuck together in aggregates is one of the reasons why the aggregation process is harmful. So you wouldn’t want an antibody to latch on to even more normal tau and cart it away.
But it appears that there is a bad actor lurking amidst the tau monomer population. Because Kayed and colleagues had originally generated TTCM2 from antibodies produced by mice when they injected oligomers into them, they had expected the TTCM2 antibodies to be selective for twisted structures like the ones in those same oligomers, which would rule out binding to normal monomers. But there were previous reports of tau monomers that were capable of “contagious” twisting into aggregates, with the “bad seed” monomers perhaps being fragments cast off of pre-existing tau fibrils.
So Kayed and his team looked to see if there was something about the 3-D structure of some tau monomers that would make TTCM2 bind to them and not to other tau monomers. They did this by subjecting some tau oligomers, fibrils, and monomers to heat and chemicals that caused them to lose their 3-D structures, and then testing to see whether TTCM2 would still bind to them.
After this forcible untwisting, TTCM2 largely stopped binding to either oligomers or monomers, while it continued binding weakly to fibrils. Meanwhile, an antibody that binds to all forms of tau worked just as well on any of these forms of tau, regardless of whether they were artificially straightened out or not. This is far from nailed down, but the authors think that the few tau monomers scooped up by TTCM2 are somehow bent out of shape and aggregation-prone, even if they haven’t yet stuck together into oligomers or other forms of aberrant tau. If that’s right, then capturing them would be a useful preemptive strike rather than unfortunate collateral damage.
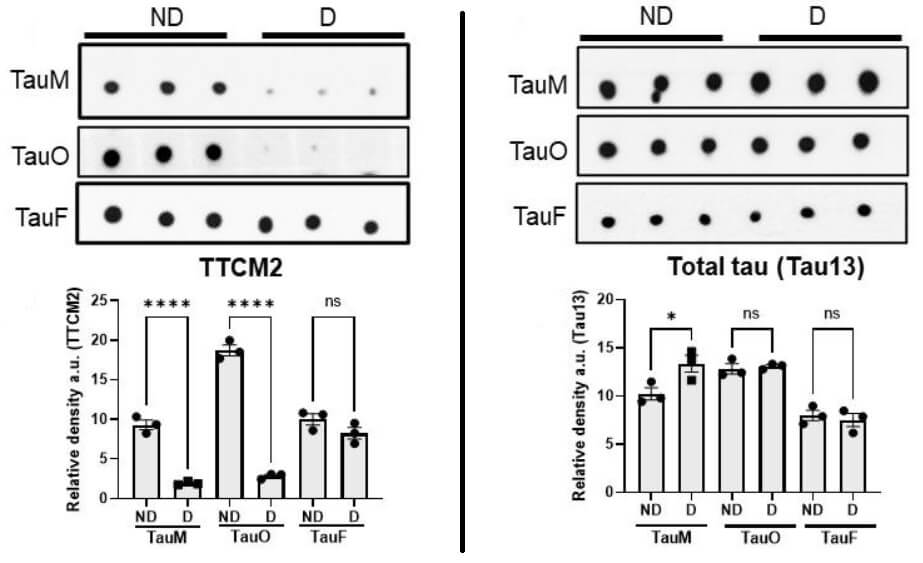
As one last initial check, the investigators did a rough-and-ready test of whether TTCM2 captures aberrant tau species responsible for the molecular templating that drives “seeding” and spread of aberrant tau from one neuron to others in its network. On this preliminary test, it did the job.
I Have Ammunition. Now I Need a Ride.
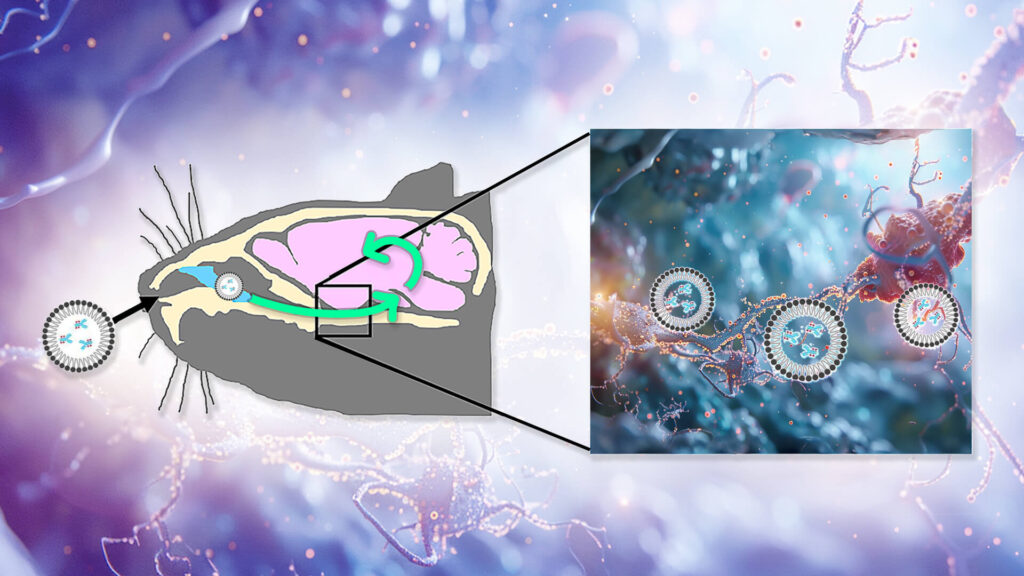
All of this suggested that TTCM2 had a lot of potential as a rejuvenation biotechnology — if they could get it to the site of the crime. To do this, Kayed and colleagues would need to get TTCM2 antibodies past the protective blood-brain barrier (BBB), into the brain, and ultimately to their targets inside neurons. To get TTCM2 into the brain, they decided to try instilling the antibody into the noses of experimental mice. In lab animals, nonhuman primates, and (in a few cases) humans, such nasal administration allows some therapeutic molecules to be swept along the channels outside of the nerves that connect the nose to the brain (including those responsible for letting you wiggle your nose and for carrying smell stimuli up to the brain) as well as along the outside of the blood vessels shared between the BBB and the inner nose.
But because therapies delivered through the nose don’t actually penetrate the nerves along which they travel, it wasn’t enough to just get the antibodies into the brain: the Kayed team still had to figure out how to get TTCM2 into afflicted neurons so they could clear out the most important depot of aberrant tau. So the team wrapped the antibodies in micelles: shells composed of fatty acids whose outward-facing heads allow them to remain soluble in the fluid bathing the brain while enveloping a water-repelling cargo inside. Micelles can often fuse with cells and release their cargo into them, which would allow them to drop TTCM2 into afflicted neurons once they got into the brain.
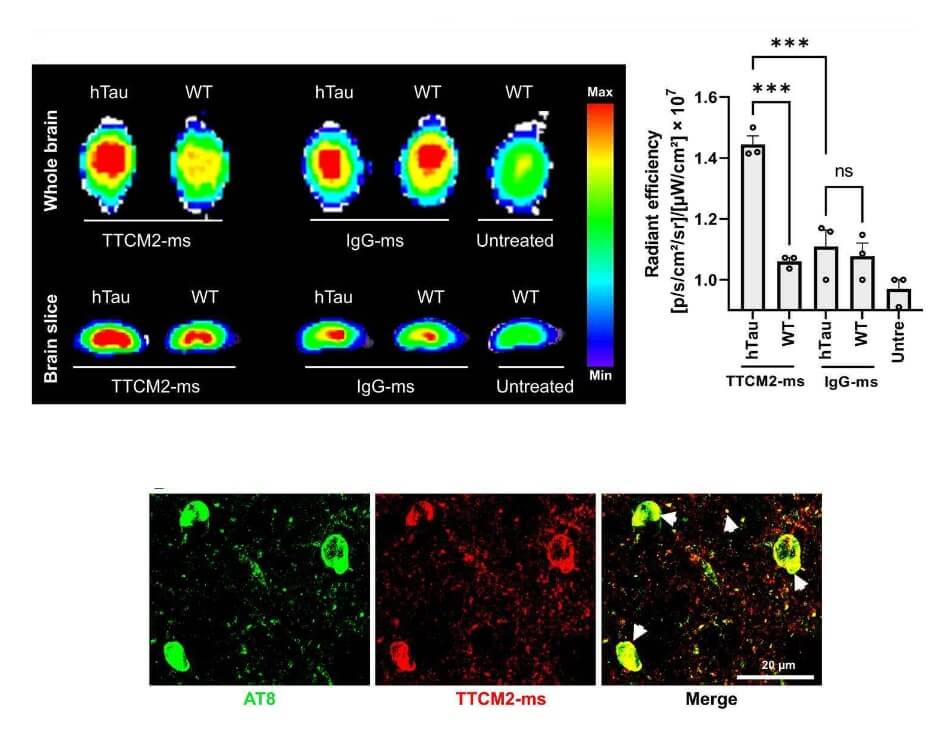
They then did a well-controlled experiment to confirm that the micelle-enveloped TTCM2 would both get into the brain and selectively latch on to areas with high tau pathology. To do this, they compared nasal delivery of TTCM2 to the brains of either aged hTau mice (which mimic the tau pathology of aging humans) or regular wild-type mice (which develop next to none of it). If TTCM2 were appropriately selective, it would bind widely in the brains of the hTau mice but hardly at all in the wild-type mice. For comparison, they also wrapped a generic antibody up in micelles and instilled it into the noses of additional groups of hTau and wild-type mice. The generic antibody shouldn’t have found anything in particular to latch onto in either kind of mouse, so unlike the TTCM2 antibodies, the generic antibody would be expected to be distributed randomly in the brain.
The antibodies reached the mice’s brains within minutes of instillation, and penetrated widely into key areas of the brain within three hours. Sure enough, TTCM2 bound widely in the brains of the hTau mice and not the wild-type; in particular, it clustered in sites of tau deposits in the hTau mice’s brains. Meanwhile, the generic antibody was loosely scattered in similar patterns across the brains of hTau and wild-type mice alike.
Clearing Tau, Restoring Cognitive Function
To this point, Kayed and colleagues had shown that TTCM2 binds to damaged tau when it’s given an easy shot at it, and that packaging TTCM2 in micelles and instilling it into mice’s nostrils would get it where it had to go. All that was prologue: the key thing was to test if it would clear those aggregates from a living brain and rejuvenate the animals’ cognitive dysfunction.
When deciding how to set up this test, Kayed and colleagues aimed for the fences. It’s all too common for scientists to begin dosing animal models of neurodegenerative aging diseases with their potential therapy before the animals have accumulated significant amounts of the target aging damage in their brains or suffered any cognitive impairment.
This might be the ideal way to give rejuvenation therapies in principle, but it sets an artificially low bar for their candidate therapy to “succeed” in this critical step. After all, if you start early enough, a therapy with even the most minimal therapeutic effect can hold off the eventual tide of cognitive decline by keeping ahead of the accumulation of damage. This can be seen in one study where researchers treated animals engineered to produce a disease-causing mutation in human tau with an antibody against it. When they gave the antibody to the mice starting when they were three months of age — before the animals had any aberrant tau pathology to speak of — the treatment greatly reduced their late-life burden of aberrant tau and protected many of their neurons from destruction. By contrast, the same antibody was unable to clear any tau pathology out of the animals’ brains when the researchers started administering it after substantial amounts of it had already accumulated in their brains.
But pharmaceutical companies don’t run multi-decade clinical trials in people who are perfectly healthy when the trials begin, and it’s not the way that most people are likely to eventually take the drug if it succeeds.
And if companies did perform their first trials of potential therapies in people with whistle-clean brains, they would need to enroll many times more people, and it would take decades to get the results. By the time they (and we) got an answer, anyone who was already in his or her 50s and older when the trials began would have already suffered decades of degenerative brain aging with no access to therapy, on a collision course with dementia.
Rather than giving TTCM2 an artificial head start by testing it in one of these unrealistic early-treatment studies, Kayed and colleagues waited until their hTau mice had already developed advanced tau pathology in their brains and were suffering cognitive impairments before beginning testing. At that point, they divided the animals into two groups, tested their cognitive function before giving them any therapy, and only then administered a single dose of TTCM2 micelles into the nostrils of one group and a generic antibody into the others’ as a control.
Two weeks after this one-shot treatment (similar to perhaps a year and a half of aging in a human), the mice given the control antibody had suffered further cognitive impairment, becoming even worse at noticing the novelty of an object the scientists had snuck into their environment than they had been during baseline testing. By contrast, the TTCM2-treated mice not only didn’t get any worse, but reversed their cognitive impairment, getting better at noticing the change than they had been the first time they were tested. Similarly, when it came to making the right choice in a simple maze, the control mice’s performance stagnated, while it substantially improved in the TTCM2 mice.

The reason for the TTCM2-treated animals’ rejuvenated cognitive performance was evident in their brains. As compared to the control animals, the brains of the TTCM2-treated mice had fewer deposited tau “tangles” and their constituents than did animals given the control antibody. They also had fewer of both larger tau aggregates and tau oligomers, and less overall warped tau protein assembled in whatever way. And in turn, the TTCM2-treated mice retained substantially more neurons in their hippocampus (an area critical for taking short-term memories and consolidating them for long-term learning). They also suffered less damage to components of neurons that are involved in forming and maintaining neuronal communication circuits — damage that was extensive in the control animals and is also prominent in human Alzheimer’s disease.
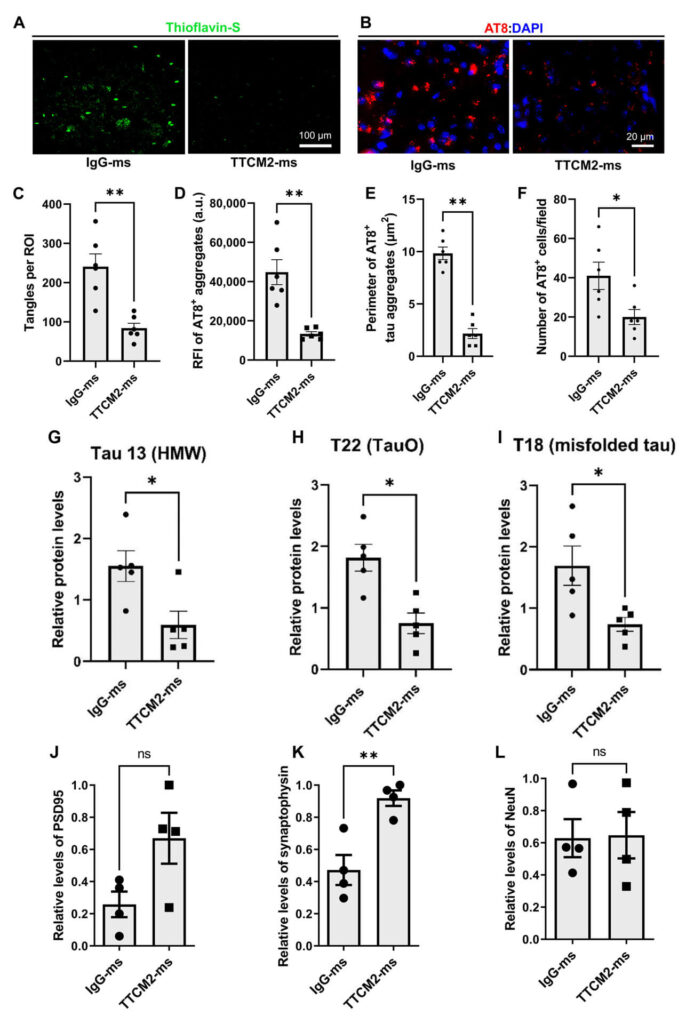
The researchers went on to show that TTCM2 specifically cleared aberrant tau inside of neurons (where it’s hardest to reach and most important to clear) as well as in the gaps between neurons through which chemical messengers jump from one neuron to the next. Capturing tau aggregates in those gaps would be expected to prevent aberrant tau in one neuron from seeding new aberrant tau in the next — and indeed, they showed that TTCM2 blocked aberrant tau from templating even more tau within the mice’s brains or when dosed on brain tissue from the brains of human AD patients.
Tag Your Garbage
Again: these remarkable results came after a single dose of TTCM2! But how exactly did the antibody do it? That is: having crossed the BBB, penetrated neurons, and bound successfully to tau oligomers and other aberrant tau species, how was TTCM2 able to make the captured aberrant tau go away? When an antibody captures beta-amyloid or aberrant tau outside the neurons, a couple of clear disposal routes are available. First, the antibody can drag its target to the brain’s resident immune cells, which then bind to the antibody via specialized receptors, internalize the bound antibody-target pairing, and send it to their lysosome for destruction. Alternatively, the antibodies can schlep captured tau aggregates out of the brain entirely, going back out across the BBB and into the circulation, where the bound pair will be destroyed by enzymes in the blood or excreted through the liver.
But once TTCM2 gets inside a neuron and traps aberrant tau in its pincers, the antibody might have been like the proverbial dog that finally catches the car. The antibody needed to be ensconced inside membrane-merging micelles to get into the neuron to begin with; once inside, it has no obvious way to make its way out again, let alone to pull its prey along with it for disposal. What could TTCM2 have done with the tau oligomers to make them disappear?
One possibility was that TTCM2 might somehow haul tau aggregates to the proteasome, a tubular protein-degrading machine that somewhat resembles the Doomsday Machine in Star Trek: The Original Series. One way that the analogy doesn’t work is that the Doomsday Machine appeared to blindly drag whatever came into its path into its maw for destruction, whereas the proteasome only degrades things that are specifically directed to it by elements of the cellular machinery.
Aware of the disposal problem for intracellular aggregates, scientists working on some of the few previous biotechnologies that targeted tau inside of neurons had designed them in different ways to direct their cargo to the proteasome. None of these had worked spectacularly well, and additionally, most of these therapies had targeted normal, functional tau instead of the aberrant tau that actually drives disease.
Kayed and colleagues had not done any special engineering to direct TTCM2 to the proteasome, but with their results in hand, they wondered if TTCM2 had somehow attracted the attention of TRIM21 to itself and its cargo. TRIM21 is a receptor inside the cell that helps degrade both viruses and some damaged or outdated proteins. It does this by binding to antibodies that have captured such miscreants and towing the whole antibody-virus complex to the proteasome. Mixing metaphors, TRIM21 is a bit like the garbage tags used in some communities, where the sanitation workers will only pick up a bag of garbage left on the curb if residents mark them with an official tag.
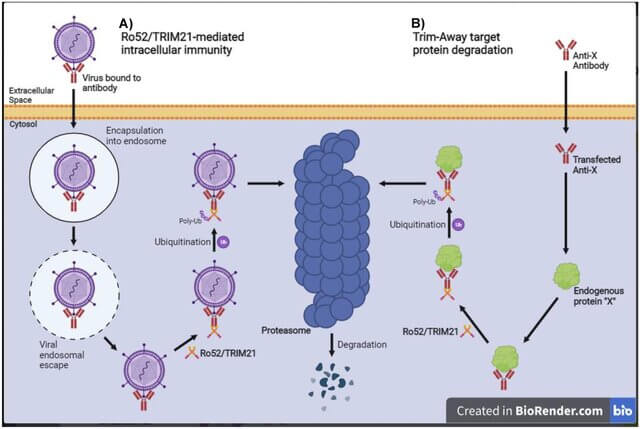
This notion didn’t pop randomly into the team’s head. There was already some evidence that TRIM21 could help clear antibody-bound tau aggregates inside cells, but it was unclear if scientists could harness that interaction to make effective longevity therapeutics. The strongest previous evidence for this phenomenon came from a study in a less faithful model of tau-driven neurodegenerative aging in which the animals were given nine doses of an antibody over the course of two months, and the authors didn’t look at tau oligomers or test whether their antibody could benefit the mice’s cognition. So it was unclear if the antibody or its interaction with TRIM21 could be used therapeutically, and the authors never tried to do so.
In this new study, by contrast, Kayed and colleagues started looking at a possible role for TRIM21 after they had already shown that TTCM2 cleared tau oligomers and other tau pathology out of the brains of hTau mice and that it could actually reverse tau-driven cognitive decline. The question wasn’t “Will it work?” but “What’s the mechanism?”.
As an initial test of a role for TRIM21, the investigators took intact slices of the brains of hTau mice and seeded them with oligomers taken from the brains of human AD patients. Within half an hour, TTCM2 bound up with aberrant tau attracted units of TRIM21 to themselves, and the tau aggregates cleared up within just one hour in this simple model.
The team then looked to see if the same thing would happen in living mice. Once again, mice were given either an inactive control antibody or TTCM2, in either case wrapped in micelles and delivered through their noses. Scientists then examined the brains of mice given either treatment at baseline, three hours later, or the next day, using fluorescent markers to see what happened to aberrant tau and TRIM21 inside their neurons. Three hours after treatment, the number of tau aggregates was about the same in both groups, but the aggregates in TTCM2-treated mice were being swarmed with TRIM21, while the aggregates in mice given the dummy antibodies were dancing by themselves.
At the 24-hour mark, little had changed for the animals given the dummy treatment: they had at least as much aberrant tau in their brains as they had begun with, and there was still little TRIM21 lining up to escort it. By contrast, the brain tissue of TTCM2-treated mice was substantially purged of aberrant tau — and at this point, TRIM21 had either disengaged (as if to say, “My work here is done”) or there were so few tau aggregates left that you couldn’t spot the few interactions remaining.
To confirm TRIM21 was actually causally involved in aberrant tau clearance by TTCM2 rather than just being in the neighborhood when it happened, the researchers repeated their brain slice work — except that this time, they blocked the cells from producing TRIM21. With TRIM21 taken off the field, TTCM2 clearly became less effective, although how much less effective wasn’t clear.
All the Boxes Ticked
As we noted earlier, many longevity biotechnologies that target aberrant tau are now in clinical trials, and others are at less advanced stages of development. But there are problems with all of them. Most of them get very little of their therapeutic molecule across the blood-brain barrier; indeed, there isn’t even a strategy for how to get the mRNA therapy targeting tau that we reviewed previously into the brain. Most would-be therapies also only target tau aggregates outside of neurons, whereas most tau aggregates are inside of neurons and do their greatest harms there. And the few tau-targeting therapies that do reach inside the brain either go the inefficient, potentially side-effect-inducing route of hoovering up healthy native tau protein in hopes of preventing it from aggregating, or are inefficient at degrading aggregates. Combined, these flaws will likely cause most of these would-be therapies to fizzle, as indeed many have already done.
Longevity Research Institute’s in-house catabody project targeting aberrant tau includes strategies to address most of these problems. Catabodies need to get as many molecules into the brain as with conventional antibodies, because each molecule of catabody can chop many pathological aggregates sequentially into pieces instead of just binding one and pulling it out of the brain. It intended to use a novel biotechnology to smuggle free antibodies into cells. And because catabodies directly destroy their targets on site, the strategy didn’t need either a way to drag tau aggregates out of the cell or a separate way to get rid of them.
Once funding picks up, we will get Dr. Sharma and colleagues back to work on the aberrant tau catabody project. But this new paper from Dr. Kayed’s lab has tackled the same challenges in different ways. They started with being the first to exploit two known delivery technologies for targeting aberrant tau: sneaking therapies into the brain via the nasal route and using micelles to get antibodies inside neurons. And they introduced for the first time TTCM2, which bears some resemblance to their previous (and promising) TOMA antibodies targeting tau oligomers, but is a novel antibody specific to the abnormal structure of aberrant tau that is easier to scale up than TOMA.
When they put the whole LysoSENS system to the test in the best available mouse model of neurodegenerative aging driven by aberrant tau, it did everything we would want it to do mechanistically. It got into the brain, dispersed across key areas of the organ, penetrated into neurons, and dragged aberrant tau kicking and screaming to its destruction. And the result was impressive: TTCM2 didn’t just slow down further cognitive decline in the mice, but rejuvenated their cognitive function, bringing it back better than it had been before they began therapy.
This would be an exciting proof-of-concept even in a world where we did not yet have AmyloSENS therapies targeting beta-amyloid, But of course, we now have those therapies available for patients with neurodegenerative aging of the Alzheimer’s type, and (clinical trials pending) we will soon have them for aging people with “normal” cognition “for their age” who have beta-amyloid in their brains. In light of the evidence that having a lower burden of aberrant tau in itself makes donanemab (Kisunla®) and lecanemab (Leqembi®) more effective rejuvenation biotechnologies, these early data with TTCM2 may be the dawn chorus of a day free from AD and other neurodegenerative aging.
* Most mouse models used to test the effects of aberrant tau on the aging brain or to test potential therapies are engineered to produce mutant forms of human tau protein that drive congenital neurodegenerative diseases in humans. Unlike in humans, these mutant mice develop severe tau tangles in the movement centers of their brains and spinal cords while still young, which physically incapacitates them. These movement problems make it extremely difficult to reliably test cognitive function in these mice, because scientists normally test cognitive function in mice by doing things like having them navigate mazes or choose between objects. So it’s difficult to convincingly show that an intervention has an effect on the cognitive function of these mutant mice because their mobility problems get in the way. On top of that, the tau pathology in these animals selectively wreaks havoc in a brain that is otherwise still young, and that therefore hasn’t been impacted by any of the other forms of cellular and molecular aging damage that ravage the aging brain. hTau mice, by contrast, accumulate tau oligomers and other aberrant tau species with age in their higher brain centers, and they become cognitively impaired later in their lives, much more like people suffering with AD and other tau-driven brain diseases — and they do not develop these florid movement disorders.