

SENSible Question: Skeletal muscles are organized into long fibers composed of many nuclei, and when a fiber is damaged, the entire fiber is often lost. It seems that if any one of these nuclei were in a senescent state and were hit by a senolytic therapy, it might result in a fiber break and pull down the entire muscle fiber with it. And muscle fibers aren’t easily replaced, and loss of muscle mass and function is already a major problem in aging, so the drug-induced destruction of muscle fibers could accelerate an aging person’s slide into disability. Is this a real risk, and if so, does it make senolytic therapies a non-starter?
This is an important question. Leonard Hayflick originally discovered cellular senescence in cells that divide: after about 40-50 cell divisions, human deep-layer skin cells would suddenly seize up and stop. Later scientists identified the erosion of telomeres at each cell division as the cause of this “growth arrest,” and researchers therefore long assumed that cells that don’t divide during the lifetime would not senesce.
If it were that simple, then there would be no reason to worry about senescence (and senolytic destruction) of skeletal muscle fibers. These are the cells whose molecular ratcheting action allows us to move our bodies in space. And they’re nondividing cells, so you might have thought they wouldn’t senesce. But there have been many reports in recent years of senescence overtaking heart muscle cells and possibly neurons and other long-lived nondividing cells. So we shouldn’t dismiss the idea that skeletal muscle fibers might senesce — and therefore be susceptible to senolytic destruction — out of hand.
That might not be too bad if neighboring cells could simply divide and replace any muscle fibers that were hypothetically torn asunder by senolytic action. But like most other cells that don’t divide by design, skeletal muscle fibers are very rarely replaced: we’ll get into the weeds on this a bit later, but the overall picture is that they are formed early in development and have to last us a lifetime. When you “build muscle” by lifting weights in the gym or on the job, you’re rarely adding new muscle fibers, but are largely increasing the size and contractile ability of the ones you already have. The physical strain of resistance exercise stimulates the fibers to build more and better ratcheting proteins within them, and it activates specialized muscle stem cells called satellite cells that repair and strengthen the stressed fibers.
Granted that there are few or no new muscle fibers coming to take the place of any that might be destroyed by senolytic drugs (until we have sophisticated cell therapy (RepleniSENS)), the question of whether muscle fibers can become senescent — and if so, what happens when they are hit with senolytic therapies — is a really important one.
So what do we know?
Mapping a “Senescence Atlas” in Aging Muscle
It’s surprisingly unclear whether muscle fibers actually become senescent or not. There are plenty of studies in which scientists report senescent cells in aging muscle tissue, but muscle is a complex tissue comprised of many different cell types in addition to muscle fibers. For starters, anyone who has ever eaten a Wagyu steak or American bacon will know that muscle tissue can be infiltrated with fat cells. Muscle tissue also contains satellite cells, as we just mentioned, and satellite cells are easy to confuse with the specialized nuclei of muscle fibers because they are localized directly on the fibers’ outer surface. The tissue also contains poorly-understood cells called fibro-adipogenic progenitor cells (FAPs), which seem to contribute to the regeneration of muscle after injury when the tissue is healthy before the insult occurs, but which also appear to drive fibrosis and fatty infiltration when the animal is aged or metabolically unhealthy to begin with. And muscle tissue is also sometimes infiltrated by different kinds of immune cells. This is true in aging, and it’s especially true after the muscle has been injured (which is central to many of the published studies on senescence in aging muscle), as immune cells rush in to clean out the debris and disinfect the wounded tissue.
So if we want to answer our critical questions, it’s not enough to just look at a sample of muscle tissue and see if there are any cells with senescent markers lurking about: we have to know whether such cells are actually senescent muscle fibers, and not senescent satellite cells, senescent FAPs, or other senescent cell types. And on top of that, we also need to rule out activated macrophages, which give misleading results on two of the most common tests for senescent cells.
The most painstaking study on senescent cells in aging muscle tissue was published last year by scientists who are now at Altos Labs, and it seemed to put fears of senescent muscle fibers — and thus the threat of senolytic-induced muscle fiber destruction — to rest. This group microdissected two different muscles (each with a different dominant fiber type) from young and old mice that had or had not been injured. No senescent cells were detected in non-injured muscles from either young or old mice using the investigators’ two primary assays, and even when they used three additional assays, only very low numbers of such cells were picked up in the old (but not young) uninjured mice’s muscle tissue.
This highlights one of the challenges of studying senescent cells: the markers scientists use to detect them have multiple functions and can be present in cells for a variety of different reasons, so you can’t definitively call a cell senescent or nonsenescent based on a single marker alone. Careful scientists therefore use multiple markers, ideally with mechanistically different connections to the senescent state — and the markers don’t always agree with each other, as in this case. What we can say at minimum is that in this study, there were no senescent cells in the microdissected muscles of young uninjured mice, and few if any even in old mouse muscle.
What was unambiguous was that after the researchers injured the same muscles, senescent cells emerged in small numbers in the young mice, and they erupted explosively after the same injury in old mice. And while these senescent cells either retreated or were cleared by the immune system two or three weeks after injury in the young mice, they stubbornly clung on in the old mice.
To check their work in “the species of interest,” the researchers also looked for senescent cells in biopsies from the thigh muscles of older human patients who had gone in for surgery. Consistent with the mouse data, there were again no senescent cells in uninjured parts of the muscle based on the assays they used, whereas senescent cells were visible in the areas around injury sites.

But to our question: what kind of cells in aging injured muscle become senescent? Do they include muscle fibers, or do all the senescent cells originate from other types of cell in muscle tissue? To answer this question, the researchers used an automated sorting system to separate out cells that showed some evidence of being senescent, and quintuple-checked them with five additional markers. They then identified the kinds of cells they had isolated in the senescent population by looking at what genes they expressed. And what they found were senescent satellite cells and FAPs (and cells derived from those two), senescent cells of the type that line the blood vessels, a variety of senescent immune cells … and no senescent muscle fibers.
Mixed Tissue, Mixed Messages
This first study was systematic in its approach to senescent cells in muscle, so you might reasonably have thought it would be the last word. If so, it would be a happy ending, since it found no senescent muscle fibers that senolytic treatments might destroy — and in executing them, potentially disrupt the function and integrity of the muscle as a whole. But then a second systematic study of senescence in aging muscle came along and threw things back into uncertainty.
Similar to the future Altos scientists before them, this second group (from the Mayo Clinic) painstakingly took apart young and old skeletal muscles from mice and then used careful methods to measure gene expression and the production of proteins by individual cells derived from five different muscle groups, along with measuring multiple other markers of senescence.
The first difference between the papers was that the Mayo researchers found significant numbers of senescent cells in the muscle tissues of old mice even if they hadn’t been injured, whereas the researchers that are now at Altos found few or none. But the more profound difference was that the Mayo group found signs of senescence in the muscle fibers themselves, not just in the non-muscle cells located in muscle tissue where the soon-to-be Altos scientists had found them.
What could explain this critical difference between these two meticulous studies’ results — and more importantly, which result best addresses the critical concern about muscle fiber destruction by senolytic therapies? One pivotal difference between the two studies was that the Mayo scientists relied heavily on assays for the expression of the gene p21, which is generally accepted as a senescence cell marker.
The future Altos group had looked for senescent cells using expression of the gene p16, which is functionally similar to p21 in that both genes encode proteins that put the brakes on cell division. Shutting down a cell’s ability to reproduce itself is one of the key modules in the senescence program, so it makes sense that these genes would be engaged in senescent cells. When the Mayo group looked at p16 expression in muscle tissue, they found that it was expressed in high numbers of cells from old animals but not those of young ones.
Those same p16-expressing cells also expressed a variety of genes that are often cranked up in senescent cells, such as genes encoding SASP factors, further reinforcing the idea that they were senescent. And the kinds of cell that expressed p16 in old animals were the same as the ones that the future Altos scientists had reported went senescent in muscle tissue with age and/or injury: non-muscle cells, such as immune cells, satellite cells, and especially FAPs. When it came to senescent cell markers other than gene expression, it was more of a mixed bag, with some markers rising in aging cells like FAPs and others not. But the general picture was still one of rising senescence in the non-muscle cells in the muscle tissue with age.
By contrast, the Mayo group found a very different pattern when they looked in an equally systematic way at p21 expression in muscle tissue. While the number of non-muscle cells in muscle tissue that expressed p16 rose with age, the number of non-muscle cells expressing p21 was low in young and old animals alike.
If the Mayo team had stopped there, the lack of an age-related change in p21 expression in the non-muscle cells might have been a curious footnote to an otherwise similar story to the earlier proto-Altos work. But then the Mayo scientists turned their attention to the expression of p21 in the entire muscle tissue — muscle and non-muscle included. And while the isolated non-muscle cells showed no rise with age in p21 expression, aging muscle tissue as a whole was expressing very high amounts of the stuff.
When the Mayo scientists looked to see which cells were producing all that p21 signal, it turned out that it was overwhelmingly coming from muscle fiber cells themselves rather than other cells in the muscle tissue. They took special care to rule out satellite cells as a source, since they are easily confused with muscle fibers because of their location on the fiber surface. They also looked for p16 expression specifically in muscle fibers; in another contrast to the non-muscle cells in the muscle tissue, they found none.
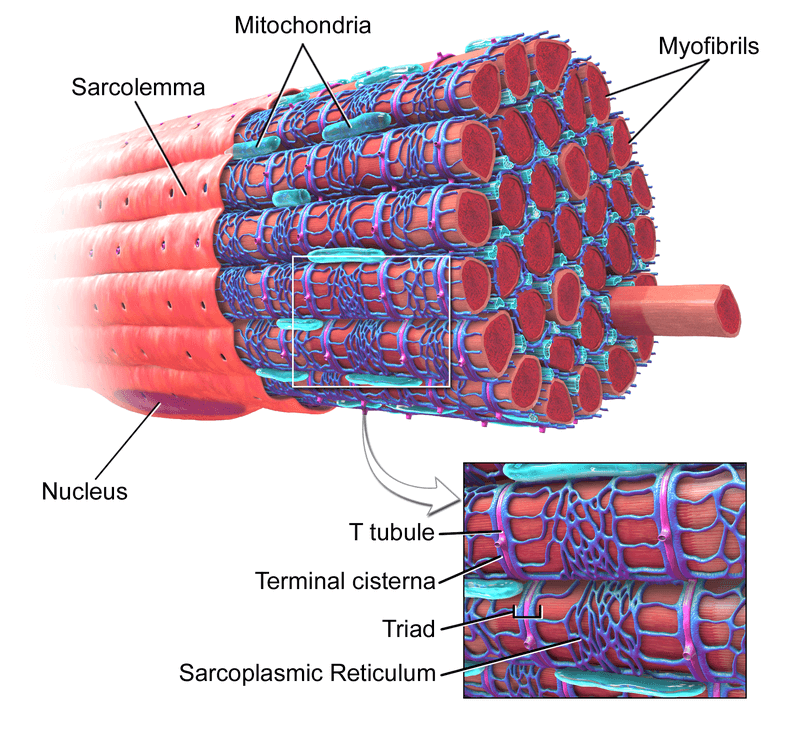
Here, we have to get a bit into the weeds about the unusual way that muscle fibers are structured. Most cells in the body are organized around a single nucleus — a protected organelle that houses the genetic code that acts as the cell’s command-and-control center. But human muscle fibers can be nearly two feet long, and if a single nucleus had to send chemical signals and microscopic biological machines running up and down the length of a cell that long, it would take so long that the muscle would not be able to carry out coordinated actions quickly enough to allow us to live our lives. So muscle fibers are instead organized with multiple specialized “nucleuses” (myonuclei) staged all along their length, each with a localized “jurisdiction” called a domain over which it’s in charge.
So when the Mayo team looked in detail at where the p21 signal was coming from, they found that both young and old mice had a few muscle fibers that each contained a small number of focal locations (presumably myonuclei) that expressed p21. But older animals had far more muscle fibers that contained intermediate numbers of hubs of p21 expression than did young animals, and only the old animals had fibers that contained very large numbers of p21 expression nodes.
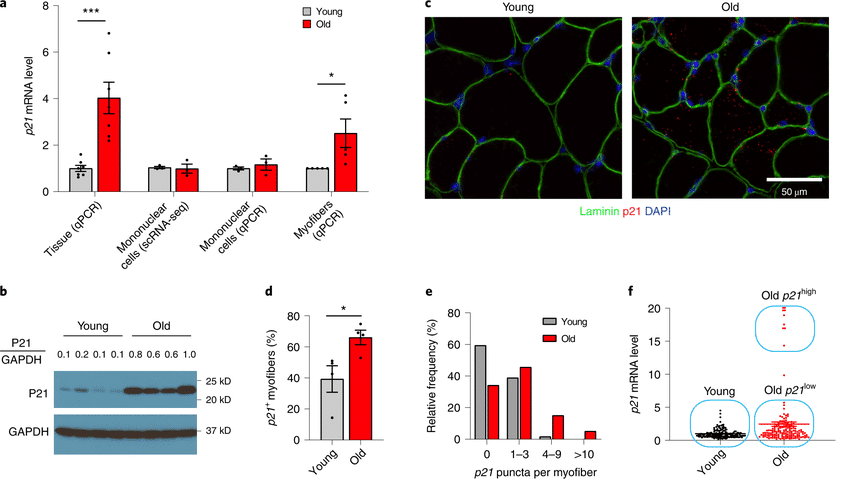
The future Altos Labs scientists mention having seen p21 “in some, but not in all, senescent cells in vivo“, but there are no actual data on p21 expression in cells provided in their scientific paper, let alone on the central question of whether muscle fibers were producing it. And unlike their exacting work in the non-muscle cells in the muscle tissue, the soon-to-be Altos group also didn’t do any analyses on isolated muscle fibers and have expressed reasonable skepticism about whether it would even make sense to call a myofiber “senescent”. So it’s not clear whether they simply missed the p21-expressing myofibers reported by the Mayo Clinic researchers because of the limitations of their methods, or if they consciously weren’t looking for them.
You Humans Are So Hard to Work With
The Mayo Clinic researchers also looked at p16 and p21 expression in muscle from young and old humans (average ages 25 and 70, respectively). Some of their results line up nicely with their mouse work. In humans as in mice, the Mayo scientists found higher levels of p21 expression in aging human muscle tissue. And high levels of either p16 or p21 expression in the biopsies correlated with lower fitness and impaired strength in the donors, suggesting a link between having a high burden of senescent cells in human muscle and age-related muscle weakening (dynapenia).
Other results from the Mayo group seem to contrast with their mouse findings. They did find that p16 expression rose with age in human muscle tissue, but while the p16 gene expression in mice had come entirely from non-muscle cells, some of the p16 signal in the human muscle tissue appeared to be coming inside the muscle fibers themselves. But this may be because unlike in in their mouse studies, the Mayo clinic scientists couldn’t look at intact isolated human muscle fibers, but were stuck working with small scattershot chunks of tissue. It’s a bit like looking at a 2-D X-ray of a person when what you really need is a 3-D CT scan. Because of this, they couldn’t be fully confident where the p16 (or p21) gene expression was coming from.
Independently, scientists from the Buck Institute and McMaster University in Canada also found that myonuclei from the muscles of old but not young men expressed p21 and several other “senescent-adjacent” genes. This group sidestepped the problem of isolating single muscle fibers by looking directly at isolated nuclei harvested from the bulk muscle tissue and then identifying the cell types from which they came based on gene expression. Because of this, their finding of increased p21 expression in human muscle myonuclei is less robust than the Mayo Clinic group’s work, but it supports the Mayo Clinic scientists’ finding that this senescence-related gene is more often activated in aging muscle fibers.
On the other hand, the Buck/McMaster team didn’t find any increase in p16 expression anywhere in aging human muscle tissue, using either of two different techniques! So we have a very messy picture on both of these markers in human tissue. And if this isn’t all confusing enough, other studies (using less robust methods than the two central studies we’ve talked about here) found no rise with age in senescent cells in human muscle, a similar lack of increase in senescent cells in the muscles of aging baboons (our close genetic cousins), and no senescent cells in unmanipulated human muscle at all.
But Are They Senescent?
Battlefield Truth
So far we’ve been asking whether muscle fibers turn senescent, with the underlying concern that if they did, then they could be hit by senolytic drugs and destroyed. The loss of a small number of senescent cells seems like a great tradeoff when the cells in question are easily replaced and fairly ‘generic’ cells like those that line your blood vessels, or the surface layer of your GI or lung, or even a working liver cell.
But a muscle fiber is quite a different sort of cell than these other cells. Unlike them, muscle fibers are not usually replaced except in response to traumatic injury — and even when acutely lacerated, the regenerative response can be stunted or incomplete depending on how the muscle is injured. This may be why even eliminating most of the satellite cells in the muscles does not accelerate age-related muscle loss in mice: satellite cells may require the inflammation and other acute responses to traumatic injury to activate a full-on regenerative response, and therefore be ill-equipped to combat the gradual degeneration of a fiber due to aging processes.* And, as has been made famous by work by Mike and Irina Conboy and others, even the satellite cells’ regenerative response to acute injury fails with age, in substantial part due to the suppressive signaling environment in the aging body.
Whatever the resolution of some of the question marks may be, it’s undeniable that the aging body’s ability to replace muscle fibers lost to cellular and molecular damage with age is grossly inadequate, and will remain so until we have advanced cell therapy and tissue engineering to overcome the limits of our inbuilt biology. Combine that with the flagging of satellite cells’ routine maintenance of most muscle fibers, and you can see why about a quarter of the fibers in the main muscle of a man’s thigh are gone by the time he is in his 70s. This loss of muscle mass and strength (sarcopenia) has terrible consequences: about 1 in 6 Americans over the age of 65 cannot lift pounds, and the numbers who can get down on their knees on their own are just as bad as that for women and are worse for men. Sarcopenia robs aging people of their independence, leading to disability, hip fractures, loss of lung capacity, and death.
And the structure of the muscle cell conjures up the nightmare scenario of a terrible friendly fire chain reaction. If the muscle fiber were to behave like other cell types that contain multiple nuclei, if a senolytic drug were to trigger “cell suicide” (apoptosis) in even one “senescent” nucleus amongst the many in a muscle fiber, it would generate a lethal wave that would provoke the self-destruction of one local nucleus after another until the entire fiber was eaten up in ruin. And because muscle fibers are organized into working bundles (fascicles) that act like coordinated strands in a “smart rope,” the consequences would be expected to extend beyond the death of the senolytic-slain muscle fiber, costing the muscle more strength than would be predicted just from its individual contribution to the fascicle’s mass.
That’s a worrying scenario for those of us who are excited by the promise of senolytic therapies. Fortunately, all the animal data refute it.
Starting when they were in late middle age, the Mayo Clinic scientists began treating their mice with the senolytic cocktail dasatinib plus quercetin (D+Q), administering the drugs in three-day cycles on alternate weeks for the following four months. The effects on senescent cells were ambiguous. D+Q appeared to reduce the amount of p16 expression in the tissue, which the researchers had previously traced back to non-muscle senescent cells (consistent with the findings from the scientists who are now at Altos) — but the difference between the treated and the “placebo” group was not statistically significant. The effect on p21 expression — which they had linked to aging senescent or senescent-like myofibers — was even less clear, with a wide spread in the level of expression after treatment in the treated animals.
A more decisive result was that the senolytics lowered the amount of expression of a variant form of p21 that Dr. Judith Campisi and colleagues have more definitively linked to senescence than expression of “garden-variety” p21. This is similar to the result of a senolytic experiment in the original report on the variant p21: treating aging mice with the senolytic drug navitoclax reduced the level of expression of the variant p21 in their tissues, but not of the standard form.
We don’t know which cells the drugs were hitting to cause the changes in expression of these various genes, and it’s possible that the Mayo researchers would have gotten clearer results if they’d been able to look at p16 only in FAPs or p21 only in myofibers. And we have no clue which cells were expressing the variant p21!
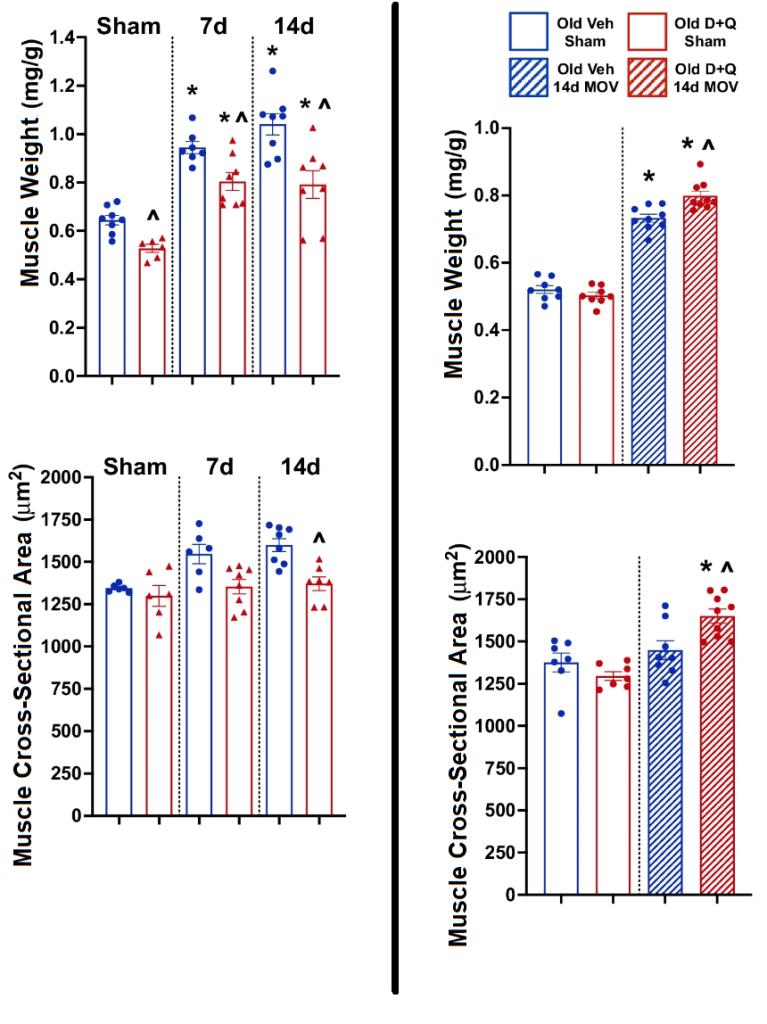
But when we get to the bottom-line question of what senolytic treatment did to the mass and function of the old mice’s muscle, we see good news all around. Not only did the senolytic-treated old mice not lose muscle, the treated animals actually sustained or restored the distribution of their muscle fiber sizes (cross-sectional area) to the same distribution seen in young mice. Senolytic-treated mice also either gained more strength or suffered less age-related loss of strength than the untreated aging animals, leaving their muscle power partway between that of old and young untreated mice. And senolytic treatment also reduced the amount of dysfunctional repair activity in their muscles.

Another study that used a form of senolytic “gene therapy” to clear senescent cells over an even longer period led to similar results, boosting the mass of most muscles and increasing the aging animals’ strength. There are several caveats to this study, however. One is that only male mice gained/preserved muscle mass thanks to this senolytic treatment: there was no apparent effect on the females. Unfortunately, the researchers did not pair the sex-specific findings on muscle mass and strength with a sex-specific breakdown on results for the destruction of senescent cells, so it might be that the gene construct itself was somehow defective in the females rather than there being a reduced benefit of actually destroying senescent cells in one sex.
Another caveat is that the “suicide gene” in this study targeted cells expressing p16, so any p21-expressing nodes in muscle fibers would have been unaffected. However, it points to a potential solution to any risk associated with killing fibers with p21-expressing nodes: senolytic strategies that specifically destroy p16-expressing cells, like Oisìn Biotechnology’s senolytic “gene therapy”. And finally, the researchers showed that the effect was not due to destroying senescent satellite cells, but it may have been at least partly due to destroying activated macrophages rather than actual senescent cells.
More support for the positive effects of senolytic drugs on aging comes from a study on the impact of senolytics on muscle gains in a model of resistance exercise in aging mice. In this study, similar to what the future Altos scientists reported, the researchers found few or no senescent cells in the resting muscles of aging mice or humans. Weight lifting in humans and muscle overload in mice caused a burst of senescent cells to erupt in the muscle, with old mice accumulating more senescent cells than young ones did from the onset and continuing to accrue more of them with ongoing overload over the next two weeks. In contrast, the burden of such cells in young animals reached a plateau after the first week of overload.
The mice gained muscle throughout the two weeks of overload, just as would happen after lifting weights in the gym. However, old mice gained less muscle than young mice did — a well-known problem that also afflicts aging humans. But when scientists treated mice with two rounds of D+Q administered 7 and 10 days into their period of overload “workout,” the old animals’ muscle gains became more like those of young ones. Notably, this happened alongside a reduction in p21-expressing cells, but it’s unlikely that the treated mice would have gained so much more muscle if the senolytics were destroying p21-expressing myofibers.
In other studies, senolytic treatment improved muscle regeneration after injury in old mice (though this may have happened more because the drugs executed excessive infiltrating immune cells rather than that they destroyed senescent ones)
The one caution in this parade of good news about senolytic treatment is that several studies find that senolytic treatment is anywhere from useless to ruinous to muscle if treatment is begun at an age when few senescent cells are in the muscle to begin with. Frequent rounds of senolytic “gene therapy” starting in early middle age had no benefit to muscle mass or strength when the mice later reached early seniority, despite the treated mice having enjoyed many other benefits to their health and a reduction in early deaths. And administering senolytic drugs to very young adult mice actually impaired muscle regeneration.
Such effects might occur because there are too few senescent cells in the muscles of middle-aged mice for clearance of them to yield a benefit, and so very few of them in very young mice’s muscles that off-target effects of the drugs on non-senescent cells outweigh the benefits of killing a tiny number of the drugs’ intended targets. And in the young mice specifically, it might also be that a transient surge of low levels of immune and/or senescent cells actually supports regeneration (as seen in young disease models). Under those conditions, clearing out the very low numbers of senescent cells in young animals would be all harm with no upside. That said, it’s important to note that even in the young mice, the senolytic treatment did not cause any harm to the animals’ existing muscles if the scientists didn’t first injure them by some other means).
One might still worry that these studies would reflect a short-term benefit of clearing senescent non-myofiber cells that would obscure the loss of a very small number of myofibers at each round of therapy, and that repeated rounds of such low-level attrition might eventually come back to bite a frequent user of senolytics. But some of these studies involved many rounds of senolytic administration over much of an old mouse’s lifespan, and there was no evidence for myofiber loss or the loss of muscle cross-sectional area in the study done by the Mayo group or in the other studies discussed here and reviewed by this author.
Molecular Uncertainty, Preclinical Therapeutic Clarity, Clinical Ground Truth
If this all seems very confusing, that’s because it is! I’ve summarized what I consider to be the most informative studies on this question, but there are many more that inform the question. While it’s clear that many of the cells in muscle do senesce, it’s not clear whether muscle fibers themselves are among them — or if so, what it means for them to be “senescent,” or whether they are the targets of the subset of senolytic therapies that have been tested in studies looking specifically at muscle. And we also don’t know which of the effects of senolytic treatment that we see are due to effects of destroying senescent cells that are locally present in the muscle versus the effects of lowering whole-body inflammation by destroying senescent cells all across the body.
What does seem clear is that whatever the answers to those questions, senolytic therapies are not inflicting any measurable harm on the muscles of older mice, and indeed are almost uniformly either preserving or improving muscle mass and function. That’s the most salient answer to our question, and fortunately it’s a reassuring one.
And the story gets better when we consider the limitations of these studies and the many ways that we could potentially improve on these results. For instance, these studies used senolytic drugs or suicide “gene therapies,” which act through brute force on the machinery of cell death and survival or execute cells based on the expression of a single gene that imperfectly reflects senescence. SENS Research Foundation scientists are working on the alternative strategy of rejuvenating or reinforcing the immune system’s ability to remove senescent cells. This ability has been honed over millions of years of evolution to integrate with the roles of senescent cells in wound healing and regeneration, and on first principles is more likely to be able to clear the “right” senescent cells at the right stage in the process: if it were not, wound healing would fail in young animals, as it does in studies of premature destruction of injury-associated senescent cells.
Additionally, we can integrate senolytic strategies with other strategies to sustain and rejuvenate muscle mass and function, such as cell therapy to replace satellite cells, or (more ambitiously) even whole muscle fibers, or (most ambitious of all) complete muscle tissue engineering; creating “backup copies” of the mitochondrial genome to prevent muscle fiber breakage at mitochondrial DNA deletion hotspots; or removing aggregated proteins in the muscle.
The final answer to our question will await the results of muscle-specific human clinical trials. And there are still molecular biology and animal studies to be done to answer mechanistic questions and fill in the gaps in the animal work. But from the available evidence, senolytic therapies seem to be not just muscle-safe, but muscle-saving.