
Neurofibrillary tangles (NFT — cytoplasmic inclusions composed of phosphorylated and abnormally-cleaved species of tau protein) accumulate in the aging brain, and at higher levels in Alzheimer’s disease and in vulnerable regions in a range of other neurodegenerative diseases; they are closely associated with neuronal death and with onset of clinical dementing disease. The clearance of neurofibrillary tangles and other intracellular aggregates from the aging brain is a key rejuvenation biotechnology to restore aging brain function.
The priority of a distinct therapy for the removal of tau pathology has become especially clear in light of followup studies in persons receiving the original, active beta-amyloid vaccine AN1792. On the one hand, vaccine responders’ brains exhibited a nearly complete absence of Aß pathology at autopsy, along with reduced neuronal loss, and in long-term (4.6 y) followup, a decline on the Disability Assessment for Dementia and Dependence Scale, stabilized hippocampal volume while adjuvant-only controls suffered ongoing declines, and extensive clearance of tau-containing neurites.(1-3,9) Yet narrowly cognitive benefits were limited, and more mature tau pathology (NFT and neuropil threads) appeared to be unaffected ((1-3), and see previous postings).This last finding, combined with the stronger association of NFT burden with clinical disease, recommend NFT clearance as a high-priority (and, ideally, complementary) immunotherapeutic approach.
In previous posts, we highlighted work by Dr. Einar M Sigurdsson and colleagues in targeting tau aggregates in animal models, demonstrating significant reductions in tau pathology and functional deficits. Now, Dr. Hanna Rosenmann’s group at Hadassah University Hospital, Israel, has reported even more impressive results, in a rodent model more closely reflecting human age-related neurodegenerative disease.
Rosenmann’s group were the first to report the presence of endogenous anti–NFT antibodies in the healthy elderly and AD patients, with the latter group exhibiting a prominent IgM isotype,(4) consistent with the feasibility of NFT-targeting immunotherapy, but also suggesting the possibility of an NFT-targeting immune mechanism in the pathogenesis of AD. In previous work (2) intended primarily to test the latter hypothesis, vaccination of transgenic tau mice with full-length tau potein “induced histopathologic features of Alzheimer disease and tauopathies, indicated by the presence of neurofibrillary tangle-like structures, axonal damage, and gliosis … [and] mononuclear infiltrates without demyelination in the central nervous system, accompanied by neurologic deficits”.(5) Their new work (6) was therefore an effort to successfully and safely clear tau pathology using a vaccination approach.
The investigators used two distinct tau-TG mouse models. The first, a relatively uncomplicated model, involved mice hemizygous for two tau mutant mutations associated with human disease: K257T and P301S (DM-Tau-tg). These mice begin to develop tau pathology at 6 months of age, along with severe deficits in synaptic plasticity and long-term potentiation (LTP). The second, “enhanced” model sought to both accelerate the development of NFT and to increase the likelihood of observing any risk from their new vaccine protocol by better modeling the inflammatory environment of the AD brain, by inducing experimental autoimmune encephalomyelitis (EAE, an protocol to induce brain inflammation, which is generally used as a model of multiple sclerosis) at 6–7 weeks of age.
These aspects of the protocol reflected the design of their original, worrisome study.(5) In that study, however, the animals had been immunized with full-length, wild-type recombinant human tau protein, whereas in this study (6) the immunogen was the pathological, NFT-related phosphorylated tau (phos-tau) species.
Following vaccination, anti-phos-tau antibodies were detected in serum and CNS blood vessels:
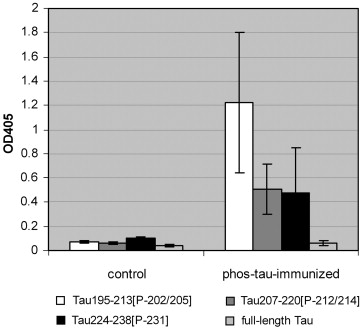
Figure 1: Anti-phos-tau Antibodies in Serum and CNS of Vaccinated Mice. (From (6))
More impressively, CNS NFT burden was reduced by ~40% following vaccination:
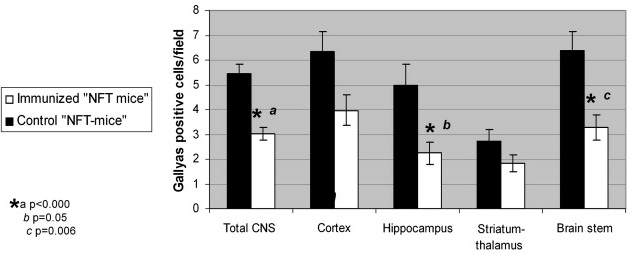
Figure 2: NFT Clearance from CNS of Vaccinated Mice. (From (6))
Importantly, while these effects were accompanied by an increase in microglial burden (which, in fact, they and others had also found to be correlated with NFT load in human tauopathies and animal models), here reassuringly the activation state of these microglia was no different from that of the pre-existing microglia in NFT-model mice, with or without the inflammatory PT “accelerant”. Moreover, “no encephalitogenicity (free of clinical neurological deficits, of adverse effects on brain inflammatory cells and of axonal damage) was recorded.” And “unlike the increase in microglial burden the astrocytes were not affected by the phos-tau immunization.
There was also evidence, consistent with results from Sigurdson’s group, of both direct antibody interaction with intraneuronal tau aggregates, and of lysosomal involvement in the clearance of the targeted species:
The level of the lysosomal proteases, cathepsins D and L, was affected in the immunized mice … [But] unexpectedly this was a decrease rather than an increase. The indication for a response of the lysosomal system, together with the presence of Igs in CNS blood vessels … [suggests that] Anti-phos-tau Abs from the periphery may reach the CNS and enter the neurons either via surface receptors … or to some extent in association with membrane-bound tau or maybe by direct translocation. The Abs may then bind to intracellular phos-tau aggregates to form anti-phos-tau Abs/phos-tau complexes. Formation of such Ab-Ag complexes can be predicted from our results showing the ability of the serum from immunized mice to recognize cell structures in the CNS of the NFT mice.(6)
While phos-tau-targeting Ab interaction with intraneuronal tau aggregates is initially surprising, it could potentially be enabled by the localization of tau protein to the plasma membrane. Moreover, as recently reviewed by Wisniewski and Siggurdson,
such an outcome is supported by a study of immunization in a Parkinson’s disease transgenic mouse model with α-synuclein showing a reduction of intracellular α-synuclein aggregates. Another study has shown that antibodies against Aβ can be internalized in AD neuronal culture models of Aβ accumulation and clear intraneuronal Aβ aggregates via the endosomal–lysosomal pathway [work in Gunnar Gouras’ laboratory, reviewed in an earlier post]. Furthermore, recent evidence has shown that extracellular tau aggregates can be internalized and promote the fibrillization of intracellular full-length tau in a tissue culture model. In addition, injection of fibrillar tau brain extract into the brains of transgenic wild-type expressing mice can induce the formation of human tau into filaments, as well as the spread of pathology from the site of injection into neighboring brain regions. This type of “infectivity” of abnormal protein conformation from outside the cell has also been demonstrated for polyglutamine aggregates and is well characterized in prion disease. Aβ has also been shown to have such “infectious” properties in vivo, being able to induce an acceleration of both further Aβ and tau-related pathology. Hence, if the spread of [paired tau helical fragment] pathology in AD can occur via such a prion-like mechanism, antiphosphorylated tau antibodies would not need to enter cells to be effective.(10)
As to the paradoxical reduction in cathepsins D and L,
recent studies did show that inhibition of the lysosomal system can indeed induce degradation of tau ([citing (7) below] mediated via activation of the non-lysosomal protease, calpain ([(8) below]) … Additional studies are certainly needed to clarify … this possible “tau degradation induced by lysosomal inhibition” mechanism. … . The suggested bidirectional role of the lysosomal system (once activated and once inhibited) in the degradation of tau may also be supported by the capability of cathepsin D not only to eliminate tau as part of activated lysosomes but also to proteolyse tau to fragments favouring tangle formation and to enhance its phosphorylation, especially when the cathepsin is released from the lysosomes and reaches the cytoplasm …
Finally, their responsibly-optimistic conclusion:
[Sigurddson’s previous work] used a different peptide immunogen and protocol (more tolerated), demonstrating its efficacy in reducing NFTs. We demonstrate here, in addition to the efficacy of our three phos-tau peptide immunogens against the NFT pathology, that our vaccine, tested under conditions aimed to detect neurotoxicity, is free of adverse effects.
The robust anti-NFT effect and the lack of encephalitogenicity in NFT mice immunized with phos-tau peptides, even though CFA with PT was included in vaccine, point to their anti-NFT therapeutic potential.(6)
These are indeed promising findings, and a clear advance in this key element of a comprehensive panel of rejuvenation biotechnologies, particularly but not exclusively for the preservation and restoration of youthful brain structure function.
References
1. Rodrigue KM, Kennedy KM, Park DC. Beta-amyloid deposition and the aging brain. Neuropsychol Rev. 2009 Dec;19(4):436-50. Epub 2009 Nov 12. Review. PubMed PMID: 19908146; PubMed Central PMCID: PMC2844114.
2. Lemere CA, Masliah E. Can Alzheimer disease be prevented by amyloid-beta immunotherapy? Nat Rev Neurol. 2010 Feb;6(2):108-19. PubMed PMID: 20140000; PubMed Central PMCID: PMC2864089.
3. Masliah E, Hansen L, Adame A, Crews L, Bard F, Lee C, Seubert P, Games D, Kirby L, Schenk D. Abeta vaccination effects on plaque pathology in the absence of encephalitis in Alzheimer disease. Neurology. 2005 Jan 11;64(1):129-31. PMID: 15642916 [PubMed – indexed for MEDLINE]
4: Rosenmann H, Meiner Z, Geylis V, Abramsky O, Steinitz M. Detection of circulating antibodies against tau protein in its unphosphorylated and in its neurofibrillary tangles-related phosphorylated state in Alzheimer’s disease and healthy subjects. Neurosci Lett. 2006 Dec 20;410(2):90-3. PubMed PMID: 17095156.
5: Rosenmann H, Grigoriadis N, Karussis D, Boimel M, Touloumi O, Ovadia H, Abramsky O. Tauopathy-like abnormalities and neurologic deficits in mice immunized with neuronal tau protein. Arch Neurol. 2006 Oct;63(10):1459-67. PubMed PMID: 17030663.
6: Boimel M, Grigoriadis N, Lourbopoulos A, Haber E, Abramsky O, Rosenmann H. Efficacy and safety of immunization with phosphorylated tau against neurofibrillary tangles in mice. Exp Neurol. 2010 Aug;224(2):472-85. Epub 2010 May 28. PubMed PMID: 20546729.
7: Wang Y, Martinez-Vicente M, Krüger U, Kaushik S, Wong E, Mandelkow EM, Cuervo AM, Mandelkow E. Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum Mol Genet. 2009 Nov 1;18(21):4153-70. Epub 2009 Aug 4. PubMed PMID: 19654187; PubMed Central PMCID: PMC2758146.
8: Zhang JY, Peng C, Shi H, Wang S, Wang Q, Wang JZ. Inhibition of autophagy causes tau proteolysis by activating calpain in rat brain. J Alzheimers Dis. 2009 Jan;16(1):39-47. PubMed PMID: 19158420.
9. Vellas B, Black R, Thal LJ, Fox NC, Daniels M, McLennan G, Tompkins C, Leibman C, Pomfret M, Grundman M; AN1792 (QS-21)-251 Study Team. Long-term follow-up of patients immunized with AN1792: reduced functional decline in antibody responders. Curr Alzheimer Res. 2009 Apr;6(2):144-51. PubMed PMID: 19355849; PubMed Central PMCID: PMC2825665.
10: Wisniewski T, Sigurdsson EM. Murine models of Alzheimer’s disease and their use in developing immunotherapies. Biochim Biophys Acta. 2010 Oct;1802(10):847-59. Epub 2010 May 13. PubMed PMID: 20471477; PubMed Central PMCID: PMC2930136.