

Short summary: A recent study claimed to find that metformin rejuvenated cognitive function in aging monkeys and lowered biological age on a nonhuman primate biological age clock. The details make the result unconvincing.
Serious scientists have championed the diabetes drug metformin as a potential longevity therapeutic for more than two decades, but more careful scientific testing of the hypothesis over the last ten years has soured many critical thinkers on this use of the drug. Notable evidence against the idea include that metformin failed to extend lifespan in rodents in the National Institute on Aging (NIA)’s Interventions Testing Program and other well-done lifespan studies; the debunking of the human epidemiological study that appeared to show that people with diabetes who took metformin lived longer than nondiabetics who didn’t; and a lot of other data covered in posts in this blog (see the links at the bottom of this post).
But interest was rekindled this September by a new study of metformin in aging cynomolgus monkeys, published in the prestigious journal Cell. The authors claim to have shown that metformin preserved brain structure, “enhanced” cognitive function, and slowed biological aging by more than six years after less than three and a half years of treatment, as measured on a new species-specific aging clock. So like gamblers who have lost their shirt but can’t stop themselves from playing, the conviction that this time it’s different has set into the longevity community.
The first and most obvious thing to note here is that if metformin really did effect a six-year slowing of biological aging that could be translated into humans, we would have a clear signal of it somewhere in the existing human clinical trial and epidemiological data, and presumably in the rodent lifespan studies. And yet — as I’ve laid out in previous posts on metformin — well-designed studies show no effect on outcomes important to aging beyond its effects on diabetes.
The second thing to note is a reminder of what the question is. The question is not, “Does metformin benefit people (or monkeys) with diabetes?” — we already know that’s true. It’s not even “Does metformin benefit people (or monkeys) with prediabetes?” — that’s also been proven, in large randomized controlled trials. For instance, in the Diabetes Prevention Program (DPP), 3,234 volunteers with prediabetes were assigned to receive metformin, a lifestyle program, or both (or placebo). The DPP showed that in less than three years, metformin cut the risk of developing diabetes by almost a third — although a very minimal lifestyle program (a low-fat diet, 150 minutes of weekly exercise (including walking), and an average weight loss of 12 pounds) was more than twice as effective.
No: we’re asking whether metformin targets fundamental aging processes, which would benefit aging people regardless of their diabetes status, in addition to having specific benefits for people suffering with the disease. So is that what the new nonhuman primate study showed? To answer that question, let’s dig into what they report about the metabolic status of their monkeys.
The Weight of the Evidence
The study centers around middle-aged male cynomolgus macaque monkeys, who were either given metformin in their drinking water at a dose similar to what diabetic humans take or untreated drinking water. (The monkeys were not old or “elderly,” as the scientific paper and the press coverage have it). The metformin trial lasted 1,200 days, which is a similar chunk of a cynomolgus monkey’s lifespan as ten years in a human.
The monkeys in the metformin trial began the study at a body mass index (BMI) of 28, which is in the “overweight” category, and remarkably, the control animals ballooned to BMI 32 (obese) just three months later.[1] They then continued to become more and more obese throughout most of the rest of the study before suffering an apparent age-related weight loss in the last year. And it’s safe to assume that these monkeys were not the rare cases of people misclassified as overweight or obese because of all the weight lifting they (weren’t) doing.
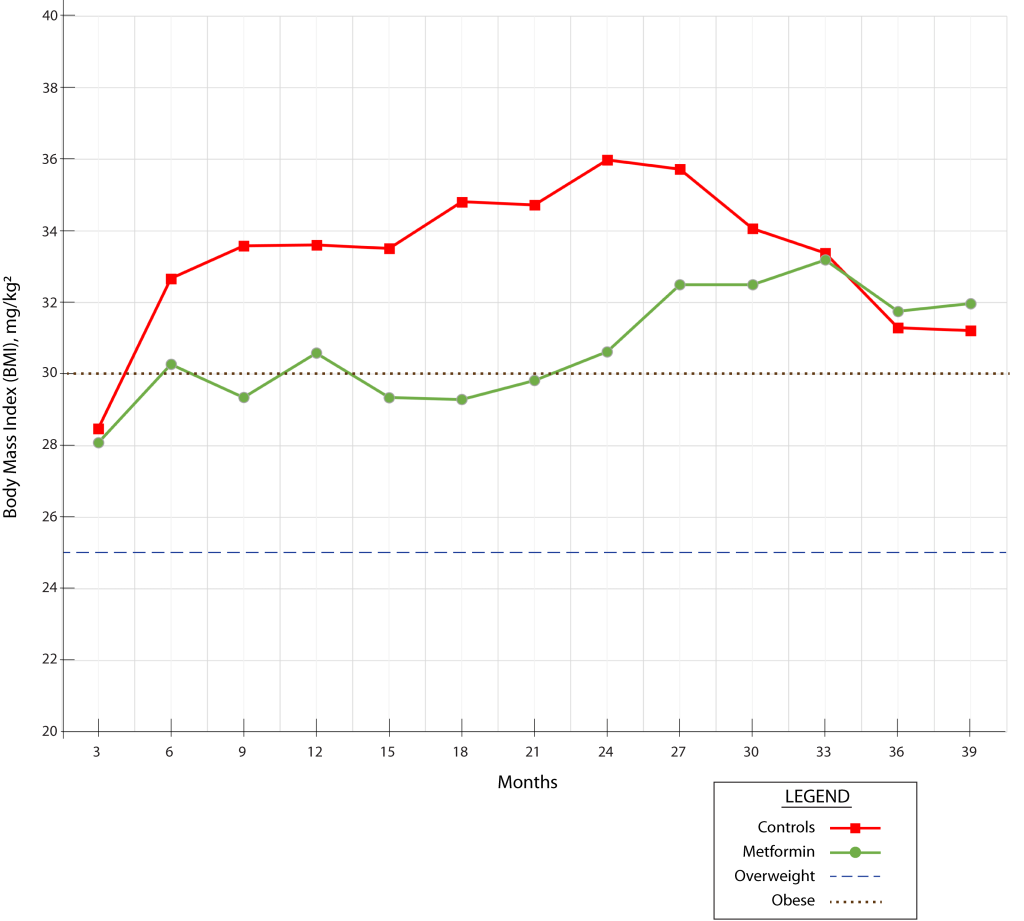
By contrast, the monkeys on metformin remained 3 to 5 BMI points lighter than the controls through most of the experiment, up until the control animals began their late-life decline. For comparison, in a 5’ 10” male human, 3 fewer points of BMI corresponds to being about 21 pounds lighter, and five fewer points amounts to 35 pounds less weight. That’s a quite profound weight-protective effect, comparable to the new GLP-1 receptor agonist drugs (Wegovy®, Zepbound®, and the like).
The authors are less than forthcoming about this critical fact: the weight data are buried in a supplemental Excel spreadsheet, and all the authors say about it is that “metformin was not associated with … a reduction in body weight” (my emphasis). This is technically true: both the metformin and the control groups gained weight over most of the course of the study. But as we’ve just noted, the metformin-treated monkeys gained a great deal less weight than the control groups did.
So the first two interrelated problems with the study are that the control animals were obese throughout almost the entire study and that metformin had such a profound protective effect against weight gain in these animals.
Why would metformin have made these monkeys so much lighter than their untreated labmates? Metformin is known to cause modest weight loss in humans, especially when it is “enhanced” by common side effects like indigestion or diarrhea. But the weight-buffering effect in these monkeys is much greater than you’ll typically see in humans on the drug.
One possible explanation for this difference is that, as we noted earlier, the researchers administered metformin to the monkeys via their drinking water, which might have put the animals off of their food due to the taste of the drug. Metformin has a very unpleasant metallic taste, which most people taking metformin barely notice because their tongues only briefly come in contact with the tablets before they swallow them. People taking Glucophage XR®, an extended-release form that is more popular than the immediate-release generics, may be even less likely to detect the taste.
However, some people who take metformin develop an unpleasant metallic taste in their mouth that they can’t escape, because a fraction of the metformin in their circulation gets continuously transported back into their mouths via their saliva. This can be a minor annoyance, or it can completely put a person off of food — as happened to my own father when he was temporarily put on the drug during a medical emergency, leading to dangerous weight loss. Obviously, if you can taste recirculated metformin in your saliva, you can certainly taste it directly when it’s dissolved in the water you’re drinking.
Whatever the mechanism, the protective effect against the substantial weight gain seen in the control animals seems highly likely to be responsible for much or all of the benefits in the metformin-treated monkeys, for reasons that have nothing to do with a direct effect on aging processes. The use of overweight and obese controls plagues aging studies, and was a fundamental flaw that hamstrung the University of Wisconsin nonhuman primate “CR” study.
In obese people, weight loss has profound beneficial effects on life and health. Even if they don’t have diabetes, obese people who undergo bariatric surgery live more than five years longer than people who don’t; that rises to nine years when people who do have diabetes go under the knife. Likewise, the new GLP-1 receptor agonists have now been shown in randomized clinical trials to lower the risk of death and other bad outcomes from cardiovascular disease and kidney disease in people with obesity and possibly slow the progression of Parkinson’s in normal-weight people. Observational studies suggest they even reduce the risk of Alzheimer’s disease in people with diabetes.
And the control monkeys were not just obese, but suffering obesity’s most direct health consequence: diabetes. The middle-aged control animals were in the early stage of prediabetes when researchers first tested their blood in the third month of the study, and by the sixth month they had progressed into full-on diabetes. During the last few months of the study, control monkeys’ fasting glucose levels surged to 180-200 mg/dL — a dangerously out-of-control level — before both groups’ glucose fell again, perhaps tied to their terminal-decline weight loss.
In addition to the control animals having persistently high fasting glucose levels even for diabetics, their values also swung wildly up and down from one testing interval to the next. This is itself a strong signal of metabolic deterioration that predicts complications and cardiovascular disease in diabetics above and beyond their average glucose level. The control monkeys’ 2-hour oral glucose tolerance tests (OGTT) were also in the diabetic range during the latter parts of the study, and they too were ameliorated by metformin.
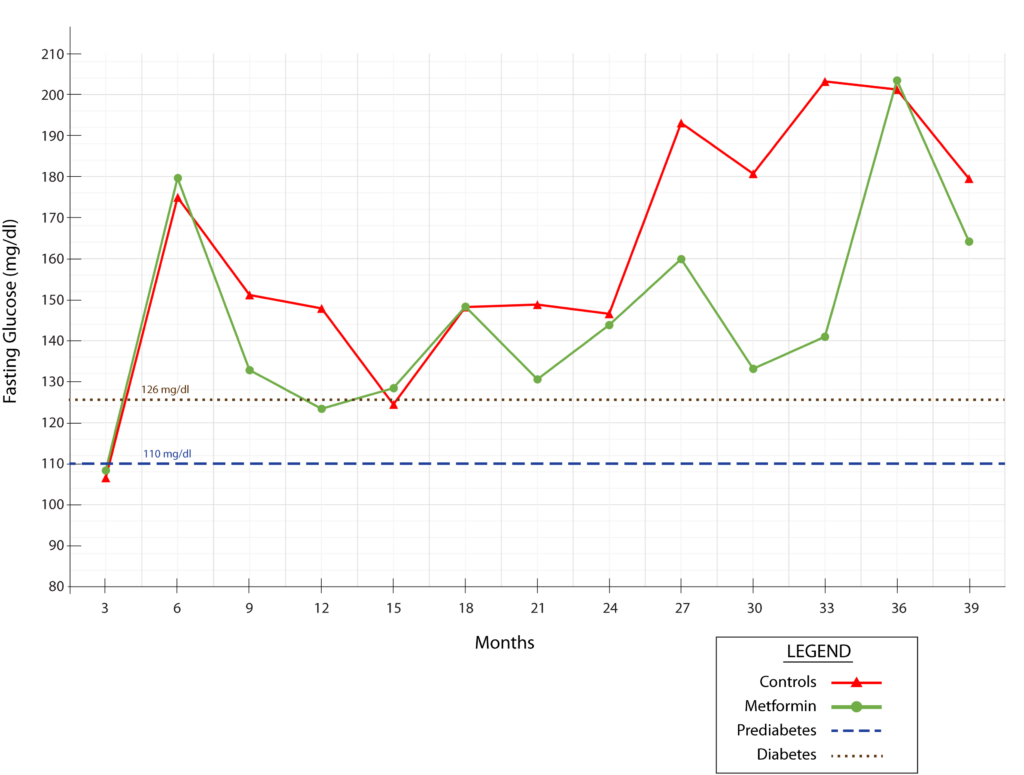
As with their obesity, the authors are not transparent about the animals’ diabetes, placing their glucose data into an oddly-constructed Supplementary Table instead of in the paper itself and only mentioning their blood glucose by saying that “metformin was not associated with compromised blood glucose homeostasis.” But the animals’ glucose homeostasis was already badly compromised even without metformin, and of course, you wouldn’t expect metformin to compromise glucose homeostasis, but to improve it — especially granted the strong weight-lowering effect of metformin in this study. And indeed, while the effect was highly variable across the trial, the metformin-treated monkeys usually had lower fasting glucose than the control animals. And the strongest effect was in the clutch: during the last third of the study, when the control group’s diabetes was at its worst.
Another opportunity where the authors failed to make the animals’ diabetic status clear was their choice to omit glucose from their urine test. Diabetes leads to spillover of blood sugar into the urine: this is the original way diabetes was diagnosed, and the very word “diabetes” comes from a Greek word relating to “passing through,” like a siphon. It is therefore routine to test for glucose in urine samples. The authors state that “we did not detect significant changes in … the physiological characteristics of urine”, and nothing is unusual in the table they label “Routine urine analysis”(which they consign to a Supplement). But that’s probably because, of all the routine urine tests they could have done, they only measured and reported specific gravity and pH!
Similarly, it is routine to measure total cholesterol as part of a “lipid panel” that includes HDL cholesterol, LDL cholesterol, and triglycerides. The ratio between HDL cholesterol and triglycerides often reveals insulin resistance, the key metabolic block in type 2 diabetes. Yet the authors measured the animals’ total cholesterol by itself.
Consistent with their diabetes, the control animals also prematurely suffered diseases of aging for which diabetes acts as an accelerant. One of the six control monkeys died of kidney failure toward the end of the trial, which would be a rare event in a healthy monkey of this age. The authors don’t state the individual animal’s age, but all the monkeys were 16-19 years old at the time this monkey died, which is roughly similar to a human aged 50-60. But diabetes is a major risk factor for kidney failure and is the leading cause of end-stage kidney disease in the United States, so the death of a control monkey from kidney failure is not such a surprise once you know that they were diabetic.
Similarly, the control animals suffered from periodontal bone loss, which was ameliorated by metformin. Lots of nondiabetic aging people get periodontal disease, but diabetes accelerates periodontal bone loss, and there is evidence that metformin protects against diabetes-associated periodontal disease.
On these grounds alone, it seems clear that most or all of the benefits of metformin on these monkeys can be chalked up to metformin’s protective effects on obesity and its metabolic consequences in Type II diabetes, rather than a geroprotective effect on the fundamental aging biology shared by all. And on top of the apparent collapse of a would-be anti-aging therapy into a weight loss drug, the remarkable results seen in these monkeys are unlikely to translate to humans because (again) we already know that metformin doesn’t deliver anywhere near the anti-obesity benefit seen in these monkeys to humans.
At this point, you could argue that further analysis of the study is moot: wherever the authors report a benefit, the answer is “but diabetes.” For instance, they say that metformin protected monkeys against age-related brain volume loss and cognitive decline. As we will see, it’s not clear that they actually showed that. But even if it were true, are these benefits because of a hitherto-unknown anti-aging effect? A simpler explanation would be that it’s because metformin was protective against diabetes, which is one of the greatest causal risk factors for dementia. But there are a lot of problems with other aspects of the study that are unrelated to conflating aging with diabetes and that make the claim of any particular benefit questionable, irrespective of the mechanism.
Never Forget Where You Came From
For starters, there were only six middle-aged animals in each arm of the metformin study, plus six additional young and young-adult monkeys (each) in some of the comparison studies and the studies by which the study scientists designed their monkey aging clocks. (We’ll clean those clocks below). You don’t have to do a statistical power calculation to know that that’s far too few animals to either derive reliable results about the effects of metformin on aging, or to build an aging clock, or to assess any metric in the study.
The authors describe the monkeys in the metformin study as “old,” but as we noted earlier, they were aged 13-16 years, which the authors themselves note is a similar point in the lifespan to a 40–50-year-old human. And the metformin trial lasted 1,200 days, which is roughly equivalent to ten human-years of treatment.
We’ve already seen that the most likely explanation for the reported benefits of metformin treatment in this study is its substantial protection against weight gain and ensuing diabetes. But it’s not actually clear whether most of the benefits of metformin in the study are even real. That’s because the authors didn’t collect any data on the monkeys at the start of the study!
That is: when you want to know the effect of an intervention on Parameter X, you should first randomly divide the subjects into two or more groups, test the subjects in each group for Parameter X, administer one group the intervention and the other a control (such as a placebo pill), and then re-test afterward to see what the intervention did.
But the authors didn’t collect any baseline data on the metformin or the regular-water control groups! The closest thing to baseline data we get is the fasting blood glucose and BMI data we graphed above, and even that didn’t start until the monkeys were already three months into the trial. All the other reported data — cognitive function, periodontal disease, brain structure, and all the biological clocks — come from after the study was over. So when you see, for instance, these data on the monkeys’ cognitive function:
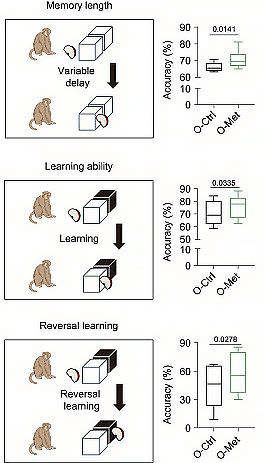
Well, the metformin-treated monkeys sure look like they were in better cognitive shape at the end of the study than their untreated labmates. But was that because of metformin? Or was that because the animals that got metformin started off the study with better brain function to begin with, and it merely carried on through to the end? Without baseline testing, there’s no way to know — especially since (as we’ve emphasized) there were only six monkeys in each group, and no documented effort at random assignment. A single Prodigy Primate in the metformin group or Cretinous Cynomolgus in the controls could have swung the group average to give the metformin group a non-pharmacological edge.
This makes it misleading for the authors to say, in the abstract, that metformin led to a “regression in brain aging” and “enhanc[ed] cognitive ability.” Both of these characterizations assert that the metformin-treated monkeys were not only better off than the control animals at the end of the study, but were actually structurally and functionally “younger” than they themselves were when the study began. But we don’t know their status at the beginning! Without baseline data, we don’t even know whether metformin slowed down the age- and diabetes-related decline in brain volume and cognitive function, let alone that it “regressed” or “enhanced” these outcomes.
Now, in some of their experiments, the authors used a poor man’s substitute to collecting baseline data: they compared the control- and metformin-treated animals’ results at the end of the study to the same metrics measured in younger animals. The idea was to show what the “normal” aging trajectory was, and then see if the drug brought the metformin-treated monkeys’ values closer to those of younger animals. For instance, here are some of their data on the brains of adolescent, young adult, and middle-aged monkeys (including the metformin-treated animals and their same-aged controls):
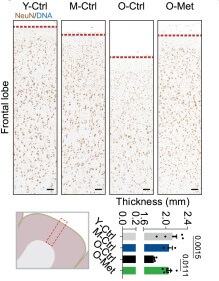
Now, to be fair, the authors couldn’t have collected baseline data for some of the experiments that they reported this way, because doing so would kill or seriously harm the animal; the brain tissue studies immediately above are an example of this. But (a) they could have collected such data from animals the same age as the metformin-treated animals at baseline; and (b) they could have collected baseline data on all the noninvasive or minimally-invasive outcomes, such as cognitive function, periodontal disease, and others.
It’s unfortunately not uncommon for researchers doing rodent aging studies to fail to collect baseline data before starting an intervention and rely on a young control group for comparison. While this practice has the same broad problems in mice as it does in monkeys, the potential for scientists to fool themselves in the process is greatly reduced in mouse studies. That’s because of the high level of control that scientists typically have over the animals before they begin the experiment. For instance, they often use a strain of mice (such as the famous C57Bl/6) that is so thoroughly inbred that their genes match up in homozygous pairs, so there’s no room for genetic variation to imbalance the two groups. And they normally acquire all the animals in a study from a single professional mouse-breeding facility like The Jackson Labs or Charles River, where all the animals are bred, housed, fed, and kept free of germs under uniform conditions, with the same climate control and the same daily lights on/lights off cycle. This again minimizes the potential for variations among the animals that pre-exist the treatment to create the illusion that the treatment had some effect that it didn’t.
Accordingly, it’s standard scientific procedure to state in a paper exactly where the investigators got their animals, at what age, how they were fed and kept, and other important details. For instance, in an important rapamycin paper by Alessandro Bitto and Matt Kaeberlein:
All lifespan and healthspan experiments were performed on 19–20 month old C57BL6/JNia obtained from the National Institute on Aging Aged Rodent Colony. A separate cohort of C57BL6/J ranging from 17 weeks to 100 weeks of age was obtained from the Harrison lab at the Jackson Laboratory for histological analysis of SFB. Animals were housed in individually ventilated cages (Allentown, Allentown, NJ) containing corncob bedding (Andersons, Maumee, OH) and nestlets. Mice were fed irradiated Picolab Rodent Diet 20 #5053 (Lab Diet, St. Louis, MO). Animals were maintained in a specific pathogen free facility within a Helicobacter spp.-free room. Mice were housed in groups (5 per cage at a maximum) and aggressive male mice were isolated to prevent fighting. Mice were acclimatized at least two weeks before onset of experiments. Mice were inspected daily, and medicated for non-life threatening conditions as directed by the veterinary staff
Now you would think that for a study involving an animal much more closely related to humans than mice are, the researchers who did the cynomolgus metformin study would have been at least equally painstaking in sourcing and documenting the background and treatment of their animals. Well, you’d be wrong:
In this study, all male cynomolgus monkeys originated from Southeast Asia. Prior to long-term metformin treatment, monkeys had no clinical or experimental background, which could impact physiological aging or heighten sensitivity to diseases. The monkeys were kept in a clean breeding room with 25°C temperature and a 12-12 hours (h) light-dark cycle at Beijing Institute of Xieerxin Biology Resource, a Laboratory Animal Care accredited facility, fully compliant with all applicable local regulations pertaining to animal experiments. Monkeys were fed three meals and had free access to water.
“[A]ll male cynomolgus monkeys originated from Southeast Asia”? That’s all the provenance we get? Were they from Cambodia, Vietnam, Singapore, Myanmar …? Did they come from a scientific animal facility with long experience caring for cynomolgus monkeys, or from other scientists’ labs, or private breeding centers focused on the exotic pet trade, or zoos, or were they captured in the wild, or …? Did they acquire all the animals from the same source, or from two or more different sources? “Fed three meals”? I presume this means daily. What were they fed, exactly? We don’t know — though we do know that whatever it was made them obese and diabetic. And so on.
And to repeat: there were only six animals in each group, and there is no record of random assignment! So what if (for instance) most of the animals came from a reputable and humane monkey facility, but two animals that the researchers put into the control group were from somewhere that gave them no cognitive or social stimulation, or a nutrient-poor diet, or where there was lead in the drinking water, or where the breeding stock suffers some genetic defect that affected their health or intelligence?
Any such differences in background between the animals assigned to each group would skew the results. This issue also emerged as a potential problem in the NIA’s nonhuman primate CR study, in which some animals were obtained from a research colony in mainland China by way of the Texas Primate Center, but others were animals previously used for military research by the US Army in Maryland. At least in that case, the NIA researchers were transparent about their monkeys’ diverse and somewhat unsavory origin; for this metformin study, we get nothing but broad geography, embracing twelve different nations plus parts of China itself. Absent baseline testing, a larger number of animals, and random group assignment, this lack of provenance makes all of their inter-group comparisons fraught.
And in addition to its potential contribution to the very strong anti-obesity effect of metformin in this study (discussed above), an additional problem with administering metformin via the drinking water is that it makes it likely that the monkeys given metformin could tell that there was something not quite right about their water, causing them to behave differently. (Yes, animals can be subject to placebo effects, and nonhuman primates are closer to human intelligence than rats or dogs). By contrast, the control group did not get any kind of placebo treatment: they just got non-treated water to drink. So we can’t discount the apparent effects of metformin being a species of placebo or Hawthorne effect, either.
A Broken Clock
As we’ve already mentioned, only six middle-aged animals (each) were either treated with metformin or used as control animals in the metformin study. To assess the effects of metformin on biomarkers of aging, the researchers sought to develop a suite of original biological age clocks for cynomolgus monkeys. For this purpose, they turned again to the same monkeys we’ve already seen them use: one group of six young (adolescent) monkeys, six of what they called a “middle-aged” group (actually a young-adult group), and the animals in the “old” groups in the metformin study (again, the “old” animals were actually middle-aged). Each of these four groups had six animals in it, and from them the study scientists designed their monkey aging clocks.
At the risk of repeating myself, the first and most obvious problem with this is that six monkeys each is far too few animals on which to build an aging clock. Thousands of individual humans from more than one cohort were involved in developing each of the well-known human age clocks, such as the original Horvath chronological age clock or the biological PhenoAge and GrimAge clocks.
And there are additional problems with the creation of the clocks reported in this study. In putting together the human age clocks, scientists first develop the clock by regressing their target data or outcome against their data set in a “training set” of thousands of adults. The data set could be anything that changes with age — DNA methylation, plasma proteins, common blood tests, etc. — and the regression target has been either chronological age (e.g., the original Horvath clock) or mortality and/or age-related disease (e.g., PhenoAge and GrimAge).
Crucially, after using an initial training set to derive their age clocks, the scientists who developed all the well-known human aging clocks then corroborated them by seeing if they predicted the same target in an independent “validation set” comprised of hundreds or thousands of different adults. By contrast, after building their cynomolgus age clocks based on a total of 24 animals (!), the researchers used them directly to assess the supposed anti-aging effect of metformin, without doing anything to validate the clocks first.
Wait: it gets worse.
To create their clock, the authors took gene expression, plasma protein, gene methylation, and other data from the three control groups discussed above: adolescent, young adult, and middle-aged. (In some cases they also appear to have used the data from the metformin-treated monkeys, which would be seriously problematic, but it’s not entirely clear because of the way the authors describe their analysis). But for the “old” (actually middle-aged) animals, the investigators used the same-age control animals they had used in the metformin trial.
This becomes problematic because they then applied the ensuing age clocks back onto the same monkeys to assess the purported anti-aging effects of metformin! It’s bad enough not to do an independent validation set analysis before using one’s age clock as an outcome for a drug study: it’s a serious problem to use the control animals’ “old” data set as the benchmark for “old” in an aging clock and then apply that clock to assess the epigenetic, transcriptomic, or other age of the very same animals that defined “old” in the clock in the first place! In other words, it’s like being lost in a hall of mirrors: the “old” monkeys look “old” because they are reflecting “old” back on themselves.

I’ll highlight just one additional problem with the authors’ cynomolgus monkey age clocks. When constructing their clocks, the researchers regressed their data onto the chronological age of the monkeys in question. There are defensible reasons to put together such a clock: the first and most famous epigenetic age clock (by Steve Horvath in 2013) was such a one. But if you’re developing a clock for the purposes of testing an anti-aging intervention, you don’t want something that just guesses how many years ago you were born. We have calendars for that purpose, and a successful longevity intervention should, in theory, have little effect on such a clock. Indeed, the Horvath clock is the weakest of many clocks at predicting long-term mortality risk and is the least responsive to potential slow-aging conditions in humans, such as exceptional long life (by current standards). By contrast, the newer mortality-based biological age clocks appropriately estimate centenarians’ biological age to be lower than their chronological age.
Instead, to test longevity therapeutics, you want a clock that measures biological age, operationally defined, for instance, as the chronological age in the general population that reflects a person’s individual risk of all-cause mortality or multimorbidity, perhaps excluding people with some individual acutely life-threatening condition. Clocks like PhenoAge and GrimAge are at least designed to do this, and do a reasonably good job at the level of a large population. But this is not the way the authors of the metformin monkey study designed their clock: instead, they trained it on chronological age, and did so with a data set that has all the problems we’ve already discussed.
Now contextualize this by considering the difficulty in knowing how to interpret the results of even the best-designed human age clocks. Since we can’t do (and don’t have the patience for) lifespan studies in humans, everyone hopes that these clocks will someday be useful as surrogate outcomes for trials of longevity therapeutics. “Damage-repair” longevity therapeutics are inherently less reliant on such surrogates than are conventional “messing-with-metabolism” geroscience, but there’s no question that a way to objectively test whether a treatment has broad anti-aging effects would be invaluable for longevity biotech.
Unfortunately, these clocks are still far from meeting the criteria we would need to use them thus. To be useful, such clocks would have to assess a person’s biological age with a high degree of both accuracy and precision. Accuracy is how close a tool’s measurements come to the real, underlying value it measures (true biological age). Precision is how repeatably the tool gives the same reading of the same measured thing, independently of how accurate it is.
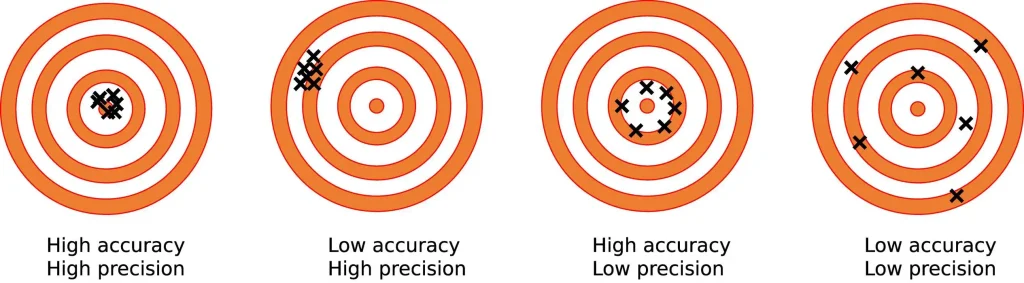
First on precision. Morgan Levine — the former Yale biomedical data scientist who co-developed some of the best epigenetic age clocks and is now with Altos Labs — has found that the actual measurement process of the underlying epigenetic markers in patient blood samples exhibits a high degree of technical noise. That is: if you take a single blood draw from a single person, split it into multiple aliquots, and run the standard measurement for epigenetic age on each of those samples, the instruments will spit back epigenetic ages that deviate from each other by anywhere from 3 to 9 years — not because of the algorithm, but because of the random variability in the measurement process itself. This noise alone would swamp a positive result from many would-be anti-aging interventions over the course of a five-year clinical trial. It also makes them useless for individuals testing themselves every six months or a year.
Moreover, while each of these clocks is predictive of chronological or biological age at the population level, the various available clocks give wildly different values from each other for any given individual. For instance, Buck Institute CEO Dr. Eric Verdin used a single blood draw to calculate his own biological age on many of the most widely-used age clocks, and got differences that varied by decades.

Karl Pfleger (an angel investor and philanthropist in longevity biotech, and the principal of the excellent resource AgingBiotech.info) had a similar experience, as have Dr. Matt Kaeberlein, Dr. Nir Barzilai and others in the prolongevist and “biohacker” communities.
Now, these human clocks were created by scientists who specialize in biomedical data and used thousands of human samples from diverse, representative samples of the population to do it — yet they are still this unreliable at the individual level. How, then, can clocks derived from a total of 18 (or possibly 24) animals of unknown provenance and that include adolescent but not genuinely old monkeys possibly be taken seriously as evidence that metformin moved the needle on degenerative aging in these animals?
Nerf Guns Against Aging
The authors also claim to show that the anti-aging effects of metformin that they purport to observe are mediated by the oxidative stress response protein Nrf2. I have some questions about that, but they’re moot: since the authors haven’t actually shown that metformin had anti-aging effects in the first place, it doesn’t matter what they think the mechanism of those phantasmal benefits might be.
To recap:
The monkeys in this study were obese and diabetic, and metformin is an established drug for managing diabetes that also led to profound weight protection in this study. Therefore, the simplest explanation for most of the observed effects of metformin in this study is that they reflect its protection from the consequences of diabetes and obesity. The animals were not even old, so it is even less likely that aging was the major driver of the health problems in these animals or that metformin was protecting them through an anti-aging effect.
Even this parsimonious interpretation may give too much credence to the results of the metformin-testing part of the study. There were too few monkeys in the metformin-versus-control experiment to have any confidence in the result, even if it was well-conducted otherwise — and it was not. There is no suggestion that the animals were randomly assigned into either group, we do not know where the animals came from or how they were treated before they came into the study, and the investigators did not collect any baseline data. From all of this, the apparent results could be nothing more than differences in the monkeys assigned to the two groups at baseline that carried through to the end of the study, irrespective of any effect of the drug.
Additionally, there was no attempt at blinding, and the animals in the metformin group may well have known something was not right with their water because of the metallic taste of metformin.
Nearly all of the problems we’ve just outlined with the metformin-testing part of the paper also undermine the credibility of the biological age clocks. On top of that, because some of the same monkeys used to construct the clocks were also used in the metformin-testing study, there is a problem of scientific circular argument built into the study. When we consider that even the best of the human biological age clocks is not yet ready for testing longevity therapeutics, these much less robust monkey clocks can’t hope to give us reliable results for testing metformin —and certainly not in a study such as the one just critiqued.
Again: metformin is a medicine with a solid safety record and proven efficacy — for people with diabetes. This monkey study supports that use — albeit weakly, for the reasons we’ve discussed. What it doesn’t support is that metformin is a longevity therapeutic.
See also the other posts in this series:
- Part 1: on the animal data on metformin and aging;
- Part 2: on some human studies on metformin, including a flawed observational study that created the illusion that diabetics on metformin actually live longer than people without diabetes;
- Part 3: on human trials of metformin to prevent or treat cancer;
- Part 4: on human studies on metformin and age-related cognitive decline and dementia; and
- Part 5: on the backstory on TAME and how it might impact the push for longevity therapeutics.
- Addendum: More Studies on Metformin and Survival
References:
[1] You may wonder if it’s valid to apply human BMI cutoffs for obesity in monkeys. This is accepted practice for Rhesus monkeys and seems to be roughly correct for cynomolgus monkeys. In one study, “Spontaneous obesity in cynomolgus monkeys was defined as BMI ≥30 kg/m2.“ In another, “The BMI of normal [cynomolgus] monkeys was 18.5‐27 kg/m2 and the BMI of the obese monkeys was greater than 27 kg/m2.” In another study testing an anti-obesity drug in “obese cynomolgus monkeys,” “Animals with body mass index (BMI) ranging from 32 to 59 kg/m2 were selected for the study.” This doesn’t seem to be universally accepted; for instance, one study “defined the reference range of BMI in healthy monkeys (20.05 to 36.73 kg/m2)” and obesity as BMIs greater than 40. But this may be an exception that proves the rule. The supposedly normal-weight cynomolgus monkeys had a liver fat fraction of 4.3 ± 0.5%, but healthy primates (human or otherwise) should have no measurable triglycerides in their hepatocytes, and 4.3% is close to the 5.0% cutoff for metabolic-associated fatty liver disease in humans.