

Short summary: One of the ways that senescent cells accumulate with age is when an initial (primary) senescent cell turns a neighboring cell senescent through its SASP secretion. As reported in a new scientific paper, SRF scientists have discovered that these secondary senescent cells are resistant to the classic senolytic drugs, allowing them to escape treatments that cause primary senescent cells to self-destruct. Through careful work, they have identified a new senolytic strategy that kills secondary senescent cells along with the ones hit by first-generation senolytics, based on their abnormal metabolism of unbound iron in a highly-reactive state. More testing is underway now to bring this “Iron Fist” strategy closer to human use.
Ever since Leonard Hayflick first reported cellular senescence back in the 1960s, scientists have been trying to understand what drives it. We now understand that senescence is a defensive program embedded in our cells to keep them from damaging the body through excessive replication that could otherwise lead to fibrosis or cancer. But when teasing out the molecular machinery that regulates the senescence program, scientists have generally studied cells in isolation from each other, or in cultures where all the cells start out non-senescent. They then expose the cells to something that puts them at risk of misbehaving (such as DNA-damaging agents, cancer-predisposing mutations, or simply being allowed to replicate themselves too much) and watch as the cells mothball themselves.
But more recently, we’ve discovered a whole new way that cells become senescent — and with it, a whole new way for senescent cells to drive aging and particular diseases of aging. It turns out that in addition to breaking down the extracellular matrix that supports cells and keeps them functional and deranging the immune system with inflammatory molecules, the SASP proteins secreted by senescent cells also shove other, non-senescent cells into senescence. If senescent cells could really do this, it would create a “domino effect:” some cells would be driven into “primary senescence” by a molecular injury of some kind, and then the SASP from those primary senescent cells would force their neighbors into secondary senescence. In turn, the secondary senescent cells would push other cells into “tertiary senescence” … and so on, and so on, into the worst nightmares of Cold War generals.

If senescence really is “contagious” in this way, it could put the foot down hard on the accelerator of senescent cell accumulation with age, as each senescent cell spreads its pathology into its neighbors and beyond. The nightmare scenario came to life in a study that found senescent cells erupting in the leg muscles of young mice a couple of months after scientists injected senescent cells into their bellies. The mice were so young that there were otherwise no senescent cells detectable in any of their tissues, and littermate controls that they injected with non-senescent cells suffered no such infestation. So secondary senescence seemed to be the only explanation for this surge of senescent cells far from where scientists had injected the original senescent cells. It could also help explain the gob-smacking results of animal senolytic studies, since under secondary senescence every senescent cell ablated would prevent several other senescent cells from forming.
But in order to put the discovery of secondary senescent cells to use in developing therapies to combat aging, we need answers to two major scientific questions. First, how far could this domino effect really spread — and thus, how much of the burden of senescent cells riddling aging tissues are the product of secondary senescence? Second, were secondary senescent cells different from primary senescent cells in some way, and would that require new ApoptoSENS strategies to destroy them?
These were the questions that SENS Research Foundation postdoctoral fellow Dr. Tesfahun (Tes) Admasu set out to answer, with help from SRF’s Dr. Amit Sharma and his lab.
Dilution Delusion
One reason to be skeptical about a secondary senescence domino theory was that several groups had previously reported that secondary senescent cells’ SASP was much less effective at propagating senescence to non-senescent cells than was the SASP from primary senescent cells. If that were so, then the problem would be self-limited. One cell would be exposed to injury or replicate for too long, and go senescent. That newly-senescent cell would turn a second cell senescent — but the signal from the secondary senescent cell’s SASP would be too weak to transform yet more cells into senescence. In this happy scenario, the chain reaction would remain subcritical and soon run itself down.
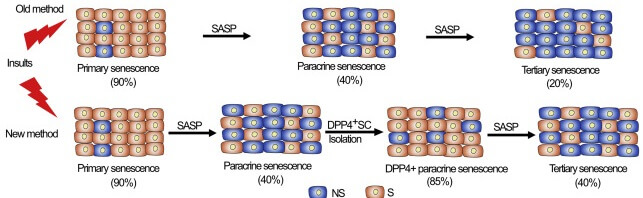
But Dr. Admasu realized that there was a problem with these experiments. When scientists tested the ability of SASP from secondary senescent cells to force nonsenescent cells to turn senescent, they were unintentionally diluting it as a function of their experimental design. Researchers would start by exposing nonsenescent cells to some stressor to create a population of primary senescent cells. They would then siphon off some SASP from these cells, expose a second population of nonsenescent cells, and see how many became secondary senescent cells. Then they would repeat the exercise, taking the SASP from this secondary senescent cell culture and using it on a third population of nonsenescent cells to see if the secondary senescent cells’ SASP could repeat the trick.
But here was the problem. The stressor that turned the original nonsenescent cells into primary senescent cells would convert a very high percentage of the original cells into senescent cells — in the ballpark of 80%. But the SASP from these cells would only convert about 40% of the second population of cells into secondary senescent cells. Because only about half as many cells in the “secondary senescent” cell population were actually senescent (and thus producing a SASP) as compared to the percentage of senescent cells in the “primary senescent” cell population, the secretions coming off of the secondary senescent cells in the “secondary senescent” cell population were effectively being diluted by the much higher number of nonsenescent cells in the culture. And that made their SASP look far less effective at switching more cells into zombie mode.
Dr. Adamsu corrected this error by using a previously-reported senescence cell surface marker to pluck primary and secondary senescent cells out of the mixed populations of cells in which they were embedded and make a real apples-to-apples comparisons between SASP from similar concentrations of primary and senescent cells. And sure enough, SASP from secondary senescent cells was just as effective at turning yet more cells senescent as was the SASP from primary senescent cells.
So yes: the threat of a catastrophic chain reaction was real. The question was, what could we do about it?
When the Bad Guy Can Dodge Bullets
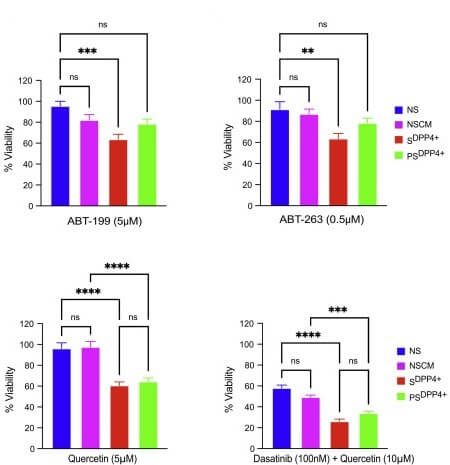
The obvious answer would be to treat aging mice or people with senolytics, and mow down secondary senescent cells along with the primary ones. But Dr. Admasu had a suspicion that this might not work. We’ve known since the first discovery of senolytic drugs that different senolytics vary in their ability to destroy different senescent cell types, depending on the cell type of origin and (as later studies would show) what forces induced senescence in a given cell. Since scientists had identified all previous senolytic drugs by testing their killing ability against primary senescent cells, it was critical to know whether these drugs would also be able to destroy secondary senescent cells in the process — or not.
Bad news: it was a ‘not.’ SRF researchers tested the effect of four of the most well-established senolytics against primary and senescent cells: Navitoclax (AKA ABT-263), Venetoclax (ABT-199), quercetin and dasatinib (the two halves of “D+Q”). Just as you would expect, the senolytic drugs had no effect on nonsenescent cells (NS) or on nonsenescent cells treated with cell medium siphoned off of other nonsenescent cells (NSCM — a control for the secondary senescent cells), while killing off substantial numbers of primary senescent cells (SDPP4+). But when the SRF team treated secondary senescent cells (PSDPP4+) with the same drugs at the same concentrations, they were either no more likely to die than nonsenescent cells, or appeared to be partially resistant to the drug. For most of these drugs to kill a meaningful number of secondary senescent cells requires concentrations of the drug that are no longer selective for senescent cells, but are toxic to senescent and nonsenescent cells alike.
Secondary Senescent Cells’ Secret
What is it that allows secondary senescent cells to dodge these senolytic bullets? To find out, SRF scientists compared which genes and proteins were ramped up or turned off in primary and secondary senescent cells as compared to each other and nonsenescent cells. The first thing they looked at was the levels of proteins from the BCL-2 family, since these proteins are known to be one of the keys to (primary) senescent cells’ ability to resist cell death. Because of this, many of the first-generation senolytic drugs work by inhibiting the activity of these proteins. By depriving (primary) senescent cells of the thing that allows them to resist programmed death, these senolytic drugs unleash the programmed cell death machinery and the (primary) senescent cells loses its battle and self-destructs.
But while SRF scientists’ tests confirmed that primary senescent cells produce high levels of all the major players in the BCL-2 family, secondary senescent cells’ BCL-2 levels were essentially indistinguishable from those of nonsenescent cells.
That explained secondary senescent cells’ resistance to at least some of the classic senolytic drugs. But if BCL-2 wasn’t responsible for keeping secondary senescent cells from self-destruction, what was? And how could that knowledge be exploited to purge aging tissues of these resistant menaces?
Digging further into the genes expressed and proteins produced by secondary senescent cells, Dr. Admasu and colleagues discovered many specific differences whose significance they have not yet puzzled out. But there was one alluring signal that they hadn’t expected: both primary and secondary senescent cells expressed high levels of numerous genes involved in iron metabolism. Despite that, there was increase in their expression of ferritin, the main form in which the body safely stores iron for future use. So what were the senescent cells up to by shuffling and transforming iron so frenetically?

SRF scientists’ tests soon confirmed that senescent cells accumulate very high levels of iron. Other scientists had previously reported this in primary senescent cells; the SENS team extended it to secondary senescent cells as well. And when they looked more carefully, they found that senescent cells were not hoarding all that iron in a safe, protein-bound, nonreactive form, but as highly reactive ferrous iron in its free or “labile” form (meaning not bound to protein). This “labile iron pool” is usually only present at low levels in the cell, with labile iron atoms dynamically entering and exiting the pool as iron is shuttled around and partakes in various biochemical reactions. To see that senescent cells were amassing such high levels of labile iron was striking.
And then they found something even more provocative. Both primary and secondary cells appeared to be on the knife’s edge of a specific form of programmed cell death called ferroptosis. Ferroptosis is a different mechanism of programmed cell death from apoptosis, which is the form of cell death targeted by conventional senolytic drugs. In ferroptosis, cells destroy themselves by using labile ferrous iron to spark up highly reactive species of free radicals, which damage fatty molecules in the cell. Enzymes in the cell then use these oxidized lipids to generate signaling molecules that lead to cell death.
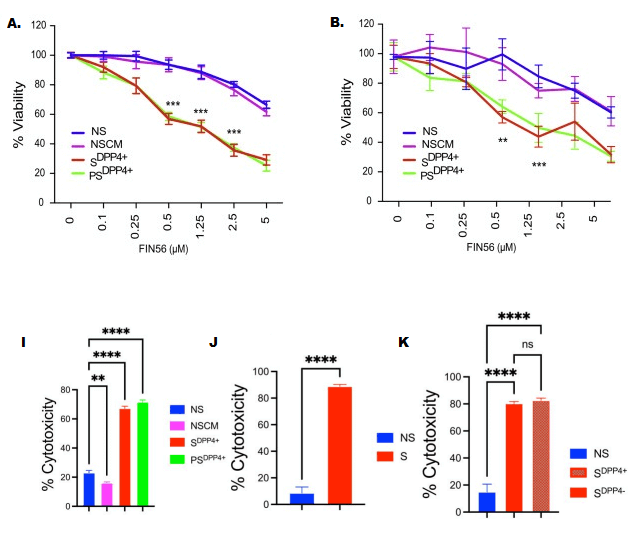
Multiple regulators of ferroptosis screamed that senescent cells were primed for ferroptotic death — and yet, the cells survived. Probing this, SRF scientists tested two drugs that are known to push cells into ferroptosis. One of these drugs (RSL3) was no more effective at killing senescent cells than nonsenescent ones. But the other (FIN56) acted like a classic senolytic drug, killing senescent cells while having little effect on nonsenescent ones. And critically, unlike the first-generation senolytic drugs, FIN56 was equally effective against primary and secondary senescent cells. And it was effective regardless of the type of normal cell the senescent cells started off as, the method by which they triggered cells into primary senescence, or whether they displayed the senescent cell marker used in the SRF scientists’ earlier isolation studies.
Dr. Adamsu wanted to be sure that FIN56’s senolytic effect was due to ferroptosis, rather than the classical form of programmed cell death (apoptosis) through which other drugs work, or sheer toxicity (necrosis), or some other mechanism entirely. So he tried dosing primary and secondary senescent cells with the drug while simultaneously administering drugs that block either ferroptosis or apoptosis. Sure enough, FIN56 was still fully effective when apoptosis was blocked, but lost its powers when SRF scientists inhibited ferroptosis.
Meanwhile, other scientists working outside of SRF independently confirmed the senolytic effect of other ferroptotic drugs, albeit only against primary senescent cells. Among them, scientists showed that both JQ1 and its more potent analog OTX01523 were senolytic. Another group of scientists showed that the ferroptotic drugs erastin, FINO2, FIN56, and RSL3 all selectively destroy primary senescent kidney tubule cells. This latter group followed it up by showing that purging old mouse kidneys of the same senescent cell type using RSL3 before transplanting them into young animals reduces the inflammation and immune cell attack by the host animal’s immune system compared to transplanting untreated old mouse kidneys.
But Still …
Ferroptosis inducers are thus a critical step up over the first-generation senolytic drugs, which failed when put to the test against secondary senescent cells. But like the first-generation senolytic drugs, ferroptosis inducers work by weakening the defenses that senescent cells throw up against a kind of self-inflicted death — a death to which they would otherwise be prone due to the corrosive effects of their abnormal metabolic state.
Because of this, ferroptosis still share one key disadvantage with the first-generation senolytic drugs, in that they only work because of differences in metabolic activity between senescent and nonsenescent cells. This can cause them to unintentionally pull the trigger on healthy cells during a moment of stress from which they could otherwise recover.
It also means that conventional senolytic drugs can destroy cells that rely more than other cells on the signaling pathways that the senolytic drug targets because of differences in their functional role in the body. That latter problem is why Navitoclax was never allowed on the market, despite showing promise against cancer. Platelets (the cells that allow our blood to clot) and neutrophils and macrophages (two important kinds of immune cells) all rely on the Navitoclax’s target BCL-2 for their day-to-day existence, so many patients taking Navitoclax suffered a crash in their platelet levels, leading to the risk of life-threatening bleeds after a cut.
The fact that senolytic drugs’ effectiveness depends entirely on differences in metabolic activity from one cell to the next is also the likely explanation for why different senolytic drugs show different effectiveness in targeting senescent cells depending on what kind of cell a given senescent cell came from and how it was pushed into senescence. That’s precisely why secondary senescent cells resist the effects of many of the most widely-used first-generation senolytic drugs.
Based on that, you’d expect that even ferroptosis-inducing drugs can vary in their senescent cell-killing ability depending on the specific characteristics of the senescent cell. Indeed, as we’ve already mentioned Dr. Admasu found FIN56 to be an effective senolytic against primary and secondary senescent cells from different original cell types, while another ferroptosis-inducing drug called RSL3 had no such effect. And beyond that, outside scientists have found that RSL3 is an effective senolytic against primary kidney tubule cells when Dr. Admasu had found it was not effective against primary or secondary blood vessel lining cells or deep skin cells.
The Iron Fist Strategy
Seeking a better solution, Dr. Admasu went on the hunt for a way to exploit the even more fundamental and distinctive characteristic of senescent cells that he had discovered: their high levels of free (unbound) iron in its chemically reduced state. Such “labile” iron can act as a catalyst to turn relatively benign hydrogen peroxide into the cell into particularly vicious hydroxyl free radicals. And he soon found one: a clever “smart bomb” that is triggered not by senescent cells’ metabolic activity, but by those labile redox-active iron atoms.
The drug is called TRX-CBI, which comprises the molecule trioxolane (TRX) linked to cyclopropylbenzindoline (CBI). You don’t need to learn how to pronounce the full words: what’s important to understand is what they do. TRX is acts as a “cage” for other molecules, keeping them from reacting chemically as they normally would. In this case, it’s caging CBI, which is a molecule that severely damages DNA and causes the cell to commit “cellular suicide” (apoptosis). But so long as it’s bound to TRX, it’s inert.
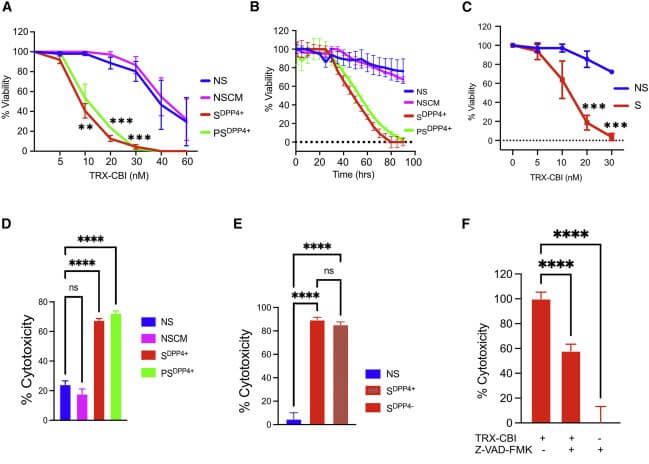
The one thing that can break open TRX and release CBI is unbound redox-active iron: such iron acts as a catalyst for the Fenton reaction, which generates especially reactive free radicals and breaks apart a critical bond between two oxygen atoms in TRX’s structure. So when TRX-CBI enters a nonsenescent cell, the near-lack of labile iron allows it to pass harmlessly through them. But when it encountenters the labile iron-rich environment inside of a senescent cell, TBI is broken open and CBI is unleashed, immediately attacking the cell’s DNA. Combined with an accompanying arc of hydroxyl radicals, CBI sets in motion a process powerful enough to obliterate the cell.

Dr. Admasu first confirmed that while free CBI itself was severely toxic to nonsenescent cells, TRX-CBI was almost completely benign to them. Then he tried caged TRX-CBI as a senolytic, and found that it performed like a dream molecule, with virtually no effect on either nonsenescent cells or a control group of nonsenescent cells treated with cell medium taken from other nonsenescent cells. By contrast, TRX-CBI was severely lethal to both primary and secondary senescent cells, including primary senescent cells of two origins and two methods of primary senescence induction.
Making it Iron-Clad
All these results (now published in the scientific journal Cell Reports and available to read for free) are extremely exciting — but so far, they’re only in cells. At this writing, the SRF team has secured p16-3MR mice for the study, a mouse model originally developed by SRF Research Advisory Board member Dr. Judith Campisi that allows for the easy tracking of senescent cell burden in mice — including its reduction by senolytic therapies. Dr. Admasu and coworkers are testing different concentrations of a DNA-damaging agent to induce high levels of senescent cells in young mice for initial testing; after that, they plan to proceed to testing in physiologically-aged mice and a model of a yet-to-be-determined disease of aging.
But we’re further along the path toward a potential human therapy than this might suggest. Dr. Adam Renslo at the Department of Pharmaceutical Chemistry at the University of California, San Francisco actually developed TRX-CBI — not as a senolytic drug, but as a potential cancer therapy. It turns out that in addition to senescent cells, a fair number of cancer cell types also gobble up iron and hold it in the labile iron pool, making them a potential target for this ingenious drug. And they’ve already done significant in vivo testing of TRX-CBI in both healthy mice and mouse models of cancer.
Animal testing with TRX-CBI has shown that tissues that aren’t burdened with senescent or cancerous cells don’t even take up particularly high levels of the drug, with the largest amounts going into healthy lungs, liver, and the envelope of connective tissue around the heart. In a mouse model of brain cancer, the normal white matter in the brain had only half as much labile iron as a human brain cancer that was growing right next to it after being transplanted into the mice In a similar study, normal mouse brain tissue had much lower labile iron than transplanted human glioblastoma cells in the same mouse.
Dr. Abdelhadi (Hadi) Rebbaa at SRF has secured funding for a project to capitalize on the insights into the iron biology of senescent cells to develop even more precisely-targeted senolytic drugs. Watch this space.
The Potential of Across-the-Board Senolytics
After so many remarkable results with senolytic drugs in aging and models of aging’s diseases, it might seem unnecessary or even unreasonable to push for new senolytics with even wider effects. Sure, we’ve always known that senolytic drugs don’t eradicate every single senescent cell in the body: every time scientists treat an animal with senolytics, some senescent cells remain — and in longer-term studies, scientists have needed to administer repeated rounds of senolytic therapies to get results. But of course, that’s also true of other “damage-repair” rejuvenation biotechnologies: even after clearing any given form of aging damage out of an aging tissue, the metabolic processes that lead to its accumulation in the first place continue and the damage accumulates anew, necessitating a future round of the same therapy.
But secondary senescent cells may be a different kind of problem precisely because they’re a different kind of cell. Different tissues accumulate senescent cells at different rates, and it’s not clear why — but it is clear that different tissues accumulate them for different reasons. For instance, the skin is more directly exposed to ultraviolet radiation than other body organs, leading to DNA damage that both promotes cancer and causes an excess of senescent cells that resist Navitoclax and require combination therapy to remove.
So are there some tissues where secondary senescent cells make up a disproportionate burden of overall senescence, or disease states that are more prone to seeding secondary senescent cells in other tissues? If so, you would never make more than a small dent in the burden of senescent cells and the havoc they wreak on the tissue, not matter how many rounds of first-generation senolytics you administered.
To date, scientists have only cleared primary senescent cells out of aging tissues, while leaving the secondary senescent cells behind. Even if secondary senescent cells behave exactly the same as primary senescent cells (which may not be the case), this could mean that specific tissues and specific diseases of aging are left untouched by the first generation of senolytic therapies.
The best way to answer these questions will of course be to see what happens when we start destroying senescent cells in large numbers for the first time with these next-generation broad-acting senolytics. SRF scientists are working to make that reality and then bring these new rejuvenation biotechnologies to aging humans.
