GlycoSENS
Repairing the Extracellular Matrix
Our bodies are comprised not only of cells, but of long-lived proteins that physically support those cells, create ordered tissue structures of them, and participate in their function. Such “extracellular matrix” (ECM) proteins are responsible for the elasticity of the artery wall, the transparency of the lens of the eye, and the high tensile strength of the ligaments, for example, as well as sustaining the function of our tissue stem cells and preventing cancerous cells from escaping their local environment to spread into far tissues.
Thus, in order for our tissues to function healthily, these ECM proteins must themselves maintain their proper structure over time. But many of these structural proteins are only recycled over the course of many decades, and others not at all.[*] Over time, they become damaged — by abnormal cross-links; by mechanical wear and tear; by abnormal mineral deposits; and by other damage caused by the metabolic and physical forces they sustain in fulfilling their duties as part of a complex living organism.
One of the most well-understood ways that ECM damage drives age-related disease, debility, and death is the stiffening of the large arteries. Several kinds of damage to the aging arterial ECM cause the arteries to lose their elasticity over time, making them less able to cushion end-target organs like the kidneys and the brain from the cyclical hydraulic battering ram of the pulsing of the blood. Without this cushion, every heartbeat damages the structures that filter our blood, threatens the integrity of vessels in the brain, and threatens the eruption of a clotting eruption from atherosclerotic plaques, leaving us at increasing risk of strokes, kidney disease, and loss of regional connectivity in the brain. Targeting these various ECM lesions for removal, remediation, or repair thus holds enormous promise to prevent, delay, and begin to reverse a sweeping range of downstream dysfunction and ill-health.
Crosslinking
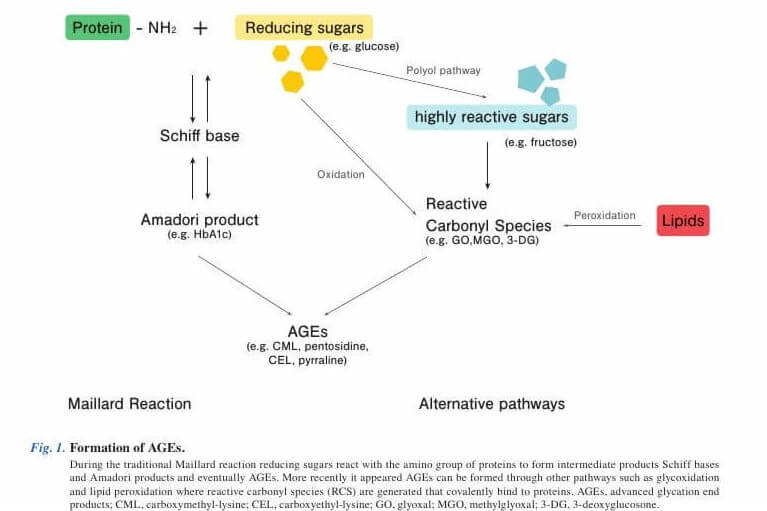
One prominent form of molecular ECM damage is crosslinking. Crosslinks act like molecular “handcuffs,” taking two previously independent neighboring proteins and binding them together, impairing their function similarly to how tying together participants’ legs in a three-legged race makes it a struggle to get to the finish line.
The main threat posed by crosslinking in the artery wall is to the strands of the protein collagen that run parallel to each other around the circumference of the vessel. When adjacent strands are crosslinked together, it prevents them from spreading apart from one another to accommodate the surge of blood sent coursing down the arterial tree by the pumping action of the heart. As more and more strands of collagen become crosslinked together over time, the blood vessels become ever more rigid, leading to a gradual rise in systolic blood pressure with age and all its deadly consequences.
Crosslinking can result from a variety of different processes. One type of cause is purely spontaneous, as reactive sugar molecules or oxidized fats in the blood react with one strand of collagen, which after a series of further rearrangements then binds to a neighboring collagen strand, crosslinking them together. Other kinds of crosslinks are formed intentionally by enzymes whose proper job is to shape and reshape the functional properties of proteins in response to other age-related changes — but such crosslinks can sometimes serve short-term needs, while becoming dysfunctional as they accumulate excessively over time.
Mechanical damage
In addition to chemical damage in the form of crosslinks, structural proteins also suffer mechanical damage, resulting from the same kinds of stresses and strains that cause automobile and airplane parts to distort or to shear. One important form of such damage in aging tissues is the fraying and breakage of structures made of the stretchy protein elastin. Whereas collagen fibrils accommodate the pulse flowing through the blood vessels by spreading outward away from each other, elastin stretches forward and snaps back into place like an elastic band. But just as an elastic band that is stretched and relaxed over and over again will gradually lose its recoil and eventually snap, so too individual fibrils of elastin in the arteries slowly wear out after decades of repeated, cyclical rounds of stretching and relaxing in response to each heartbeat, eventually causing them to fray and tear. Other drivers of damage to aging elastin include free radical attack, enzymes that degrade elastin in a failed attempt to remodel the tissue, and (in the skin) UV radiation from the sun. Similar processes appear to be afoot in the lungs.
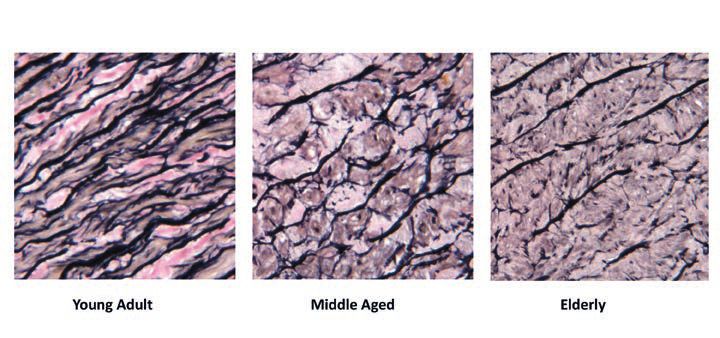
As with crosslinking, the mechanical and enzymatic degradation of elastin structures in the arteries also leaves the kidneys and brain more vulnerable to the direct pounding of the pulse, while the fraying elastin in the lungs contributes to the loss of lung capacity in aging — and especially in diseases of the aging lung.
Elastocalcinosis
In addition to its direct effects on elastin structure and function, mechanical and enzymatic damage to elastin makes it vulnerable to elastocalcinosis, the progressive process through which elastin fibrils almost literally turn to stone with age. Starting in one’s forties, arterial and other elastin becomes progressively mineralized with calcium deposits, causing the flexible elastin structure to gradually harden.
(In case you’re wondering: elastocalcinosis is a different sort of “vascular calcification” from the better-known form of calcification associated with atherosclerotic plaque. That kind of “vascular calcification” occurs on the surface of the plaque and is scored on coronary calcium CT scans, whereas elastocalcinosis occurs inside the artery wall where arterial elastin is located and accumulates in diseased and plaque-free regions of the vessel alike. The “vascular calcification” scored on coronary CT is a powerful indirect signal that a patient is at high risk of future heart attack and stroke, but is probably relatively harmless in itself, whereas elastocalcinosis is a direct causal driver of age-related arterial stiffening, rising systolic blood pressure, and end-organ damage).
One driver of elastocalcinosis is fraying of elastin, as a result of mechanical damage or enzymes induced by inflammation and oxidative stress. In youthful arteries, sites within the elastin structure that are intrinsically susceptible to unintended calcium deposits are shielded from calcium ions by an outer layer of fibrillin proteins, but damage to the elastin structure disrupts this protective coating and exposes the vulnerable core elastin. Other contributors to elastocalcinosis include unintended side-effects of metabolic processes involving growth factors and misdirected bone-mineralizing proteins.
Rejuvenation Biotechnology Solutions
The kinds of aging damage to which ECM is subject are sufficiently varied that — unlike nearly all other categories of aging damage — the various kinds of GlycoSENS molecular lesions can’t all be removed, repaired, or replaced with variations on a single underlying rejuvenation biotechnology platform. Instead, each subtype will require a more specific attack strategy.
All things being equal, one thing that makes AGE crosslinks a relatively easy target is their very unusual chemical structures, which are not found in proteins or other molecules that the body makes on purpose, including other (enzymatic) crosslinks. This should make it possible to identify or design “AGE-breaker” drugs or enzymes that can chemically cleave these aberrant crosslinks apart, without damaging normal proteins or other structures. In the 1990s and into the 2000s, scientists seemed to have found just such a molecule: a drug called alagebrium (or ALT-711), which substantially lowered tissue stiffening in aging or diabetic mice, reduced arterial stiffness in nondiabetic aging dogs, and even improved the stiffening of the arteries and hearts of nondiabetic aging rhesus monkeys, in addition to several other beneficial effects.
In humans, however, alagebrium’s effects proved to be much more limited in scope and modest in magnitude. After the false dawn of a seeming success in the first major clinical trial (likely an artifact of the way the key outcome is calculated), alagebrium proceeded to fail in a series of clinical trials for different diseases of the aging cardiovascular system, and had no effect (or a weak , limited, or unconvincing one) in trials combining it with exercise. This may be because the kind of crosslink that alagebrium severs is much rarer in humans than it is in rodents and other experimental animals, or because its mechanism(s) of action are actually not related to AGE-breaking.
Now, the search is on to develop new AGE-breakers that specifically target crosslinks that accumulate in large quantities in humans. Current research suggests that a very complex molecule called glucosepane is the single greatest contributor to total AGE crosslinks in aging human tissues. Type I diabetics’ burden of glucosepane strongly predicts their risk of cardiovascular disease, above and beyond the usual marker of diabetic control (HbA1c), and it is associated with faster progression of diabetic complications.
Therefore, glucosepane-cleaving therapeutics will likely have the biggest tissue elasticity-rejuvenating effect as compared to drugs that target other AGE crosslinks. But glucosepane is a very difficult target. For one thing, its large size and complexity may make it impossible to break with the simple small chemical molecules that are typically used as drugs. Instead of drugs, finding or engineering enzymes to break glucosepane may be a more viable approach. On the other hand, it’s possible that many of the glucosepane crosslinks in our arteries are ‘buried’ inside tightly-knit fibers of collagen where such enzymatic therapeutics would not be able to reach due to their size. Clearly, a range of possible approaches to glucosepane AGE-breaking needs to be explored.
Overcoming the fraying of the elastin structures in the arteries and elsewhere in our bodies, as well as elastocalcinosis, will require entirely different solutions. Solutions to the former may involve tissue engineering solutions, such as transplantation of new elastin structures, or wholesale replacement of parts of the large arteries. Work is ongoing in a wide range of approaches to tissue-engineered arteries, though only a subset of these strategies address the key need for an integrated elastin lamellar structure. The decellularized-recellularized tissue scaffold approach has shown promise, but as with kidney or lung transplantation, this approach relies on intact donated tissue from young people who die in accidents or of other infrequent causes of death — a solution that can’t be scaled to the entire aging population.
On the other hand, after an extended period of scientists drawing blanks when trying to come up with a strategy for repairing elastocalcinosis, a promising new approach has recently emerged and is under active pursuit (see below).
Where We Are Now
Revel Pharmaceuticals, a spinout of SRF-funded research at Yale University, is now turning its groundbreaking work on glucosepane into potential rejuvenation biotechnologies. With a ready supply of glucosepane on hand thanks to Yale’s new method for synthesizing it, Revel seized upon the emerging techniques of metagenomic libraries to look for bacterial enzymes capable of cleaving it. Revel scientists chopped up the genomes of complex, unidentified environmental bacteria without prior knowledge of what they may encode, and inserted one gene encoding one of their proteins into the genome of one uniquely-engineered lineage of the common E. coli bacteria. Each cultured bacterium then expresses one protein originally derived from the environmental bacteria, and the encoded protein — by way of the encoding bacterium — can be screened against a target. This technique allowed Revel scientists to screen the vast, uncharacterized enzymatic armamentarium of unidentified, uncultured environmental bacteria with an efficiency approaching the high-throughput methods used for conventional small-molecule drugs.

The most important fruit of this research is the discovery of bacterial enzymes that could break down glucosepane crosslinks. They are now working to further test these candidates, with the aim of modifying them into forms that would be suitable for new longevity therapeutics. (See more about SRF, Yale, and Revel under “SENS Research Foundation Research” below)
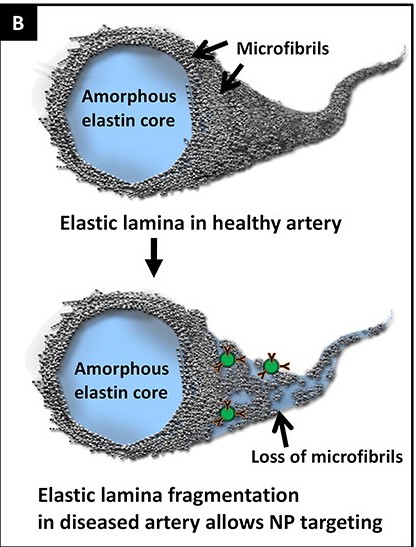
On the elastocalcinosis front, Dr. Naren Vyavahare and colleagues at Clemson University have developed an ingenious potential way to remove calcium deposits from damaged elastin and simultaneously help the body repair the underlying damage to the elastin protein. This method offers an exciting potential solution to a challenging set of problems for which no one had previously proposed a viable repair-based strategy. Now spun out as Elastrin Therapeutics and supported by seed funding from Kizoo Technology Capital, this experimental rejuvenation biotechnology is built around hollow nanoparticles whose surfaces are covered with antibodies that selectively bind to the core protein of the elastin structure.
Because the core protein is only exposed and available for binding when the outer coating of fibrillin around the elastin protein has been ruptured by enzymes or mechanical forces, the antibodies bind only to damaged elastin, while leaving healthy elastin and other proteins alone. With the antibodies tethering the nanoparticles tightly to damaged elastin, the nanoparticles release a payload of encapsulated drugs at high local concentration, while almost eliminating systemic exposure elsewhere in the body
In multiple mouse models of age-related diseases of damaged elastin, Vyavahare’s team have demonstrated that these functionalized nanoparticles selectively target damaged elastin in vivo, dissolve accumulated calcium, and facilitate elastin repair. The nanoparticles target sites of elastin calcification due to atherosclerosis, elastocalcinosis, and kidney disease; reverse abdominal aortic aneurism in several different animal models; and improve lung function and preserve lung elastin in animal models of emphysema. Once the repair of the underlying damage begins, the inflammation of the affected vasculature wanes, and immune cells stop attacking the damaged tissue, creating more favorable conditions for durable repair.
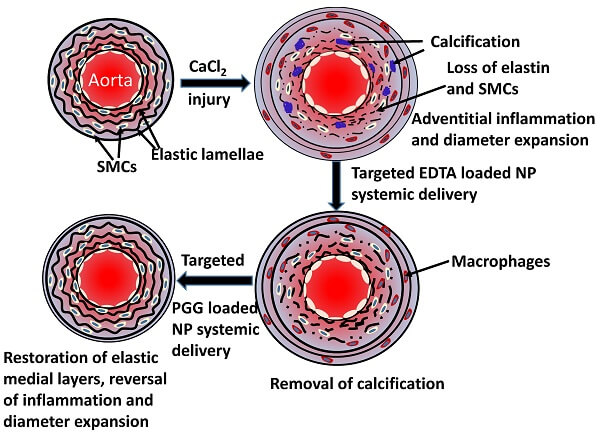
Additionally, by loading the hollow spheres with gold instead of drugs, the Elastrin team have shown that these same nanoparticles can be used to noninvasively visualize the location and extent of vascular elastin damage, creating a tool that can be used to diagnose diseases of damaged elastin, assess risk of catastrophic outcomes, and monitor the response to future rejuvenation biotechnologies, while facilitating their own and others’ ability to develop and test such therapeutics.
SENS Research Foundation Research
The glucosepane research in Dr. David Spiegel’s lab at Yale that led to the successful spinout of Revel was originally conceived and driven by Dr. Aubrey de Grey. Dr. de Grey clearly saw the need to jumpstart work on glucosepane-cleaving rejuvenation biotechnologies after the failure of alagebrium. Not only had the overall pursuit of AGE crosslink breakers stalled out, but there had never been a serious effort to target glucosepane, in large part because of how difficult it is to study and target with new therapeutics. So SRF initially funded research at Yale that was not intended to lead to any one specific group developing one specific candidate, but to generate the key reagents that scientists everywhere could use as tools. These tools would effectively break open the field, enabling researchers to potentially target glucosepane, without requiring a massive and redundant investment of time, specialized chemists, or highly specialized and expensive equipment. It ploughed the earth so that a thousand flowers could bloom.
The first challenge to developing glucosepane-targeting therapies is glucosepant’s massive, baroque structure, which exists as eight different variants within tissues depending on the orientation in space of the bonds within its structure. Glucosepane had thus never been synthesized in the lab at the time the project began, whereas many other AGE products are easily cooked up (literally) by heating simple sugars together with easily-available proteins. The lack of a ready supply of glucosepane made it difficult to test whether drugs or other therapeutic candidates would be able to cleave it, or to study its structural properties and behavior for weaknesses that could be exploited.
To address this challenge, Dr. Spiegel and his team first used SRF funding to achieve the first complete synthesis of glucosepane in the lab — an accomplishment significant enough to merit publication in the prestigious scientific journal Science.
Next, SRF also funded the Yale scientists to develop reliable antibodies that specifically bind to glucosepane. SRF had previously sponsored Dr. William Bains’ laboratory in Cambridge, which demonstrated that the antibodies commonly used to identify other widely-studied AGE are miserably non-specific — and prior to the Yale group’s work, there was no antibody specific for glucosepane. The lack of such antibodies made it difficult to determine the burden of glucosepane in aging or diabetic tissues before and after treating tissues or animals with candidate glucosepane crosslink breakers as a test of efficacy, again severely constraining research. Absent such antibodies, the only way to test candidate glucosepane-breakers would be to directly isolate glucosepane from tissues — a complex, time-consuming processes involving multiple rounds of enzymatic digestion, which most labs are ill-equipped to do. Having antibodies that are highly specific for glucosepane solves this problem, releasing a constraint on glucosepane therapeutic research.
These two tools — synthetic glucosepane, and glucosepane-binding antibodies — therefore crack open the field for the development of glucosepane-cleaving rejuvenation biotechnologies — both by Revel and by up-and-coming competitors.
Separately, we’re also funding research by Dr. Jonathan Clark at the Babraham Institute in Cambridge to both identify new high-priority crosslinks to break with future rejuvenation biotechnology, and to demystify the complex tissue-function-shaping behavior of crosslinks, as well as how the mix and locations of crosslinks change with age.
*Unlike these long-lived structural proteins, the proteins inside our cells are continuously being broken down and rebuilt to replace damaged structures and to match the level of specific proteins and organelles inside the cell with its metabolic needs at any given time. When the systems responsible for breaking down and recycling these proteins fail, they leave harmful waste material inside cells, but for the great majority of proteins, these systems operate quite well when the cell is youthful and healthy. The exceptions are aberrant proteins that the cell has never evolved the ability to degrade and thus slowly accumulate throughout life; it is for these exceptional intracellular aggregates that LysoSENS strategies are needed.
Repairing the Extracellular Matrix: Request for Proposal
The SENS Research Foundation is seeking proposals for innovative approaches to repairing age-related extracellular damage. Proposals should include a comprehensive description of the proposed approach and any supporting evidence, as well as a detailed budget and timeline for completion.