
Short summary: Several pharma companies are currently running clinical trials on damage-repair therapies targeting damaged forms of the protein tau to combat Alzheimer’s disease. But these AmyloSENS therapies only reach tau in the fluid outside of neurons, when what we need is to clear damaged tau inside of them. Fortunately, researchers are beginning to use mRNA — the same revolutionary biotechnology platform of the best COVID vaccines — to develop new LysoSENS therapies to do just that.
In the last two years, FDA has approved two AmyloSENS therapies to combat neurodegenerative aging of the Alzheimer’s type (AD), with the latest approved while this post was in editorial. These are victories on two fronts. First and foremost, donanemab and lecanemab (Leqembi®) are the first therapies ever proven in large clinical trials to put the brakes on the terrible fall into living oblivion that is AD. And on top of that, they are also the first SENS (damage-repair) therapies approved for any disease of aging.
Critics have put an outsized focus on the limitations of these results: that donanemab and Leqembi® did not cure AD in these trials, but “only” acted as parachutes to keep people cognitively aloft for longer. This is true as far as it goes — but as we’ve emphasized before, it’s an unreasonably myopic and pessimistic take on the revolution that these therapies set in motion. These AmyloSENS therapies are necessarily limited by the fact that they only remove one form of aging damage from the aging brain (beta-amyloid aggregates). By the time people reached the clinical stage at which they were eligible for these initial trials, beta-amyloid had already had decades to inflict downstream molecular and cellular wreckage in the brain, including driving aberrant tau into the neocortex and destroying a third of the neurons in the entorhinal cortex. At that point, removing beta-amyloid aggregates can prevent even more of this downstream damage from assauting the brain, but it can’t undo the non-beta-amyloid damage that the brain has already suffered.
As we’ve discussed, now that science has put this first crack in the wall of AD, there are two non-exclusive ways to leverage it to send the wall tumbling down, leading to the indefinite postponement and functional cure of AD. One is to start clearing beta-amyloid much earlier, before the downstream damage has set in. Scientists at Eisai (which makes Leqembi®) and Eli Lilly (which makes donanemab) are testing that approach right now in the AHEAD Study and TRAILBLAZER-ALZ 3 trial, respectively.
The other is to remove and repair the downstream damage directly, after it has occurred, starting with the damage most directly inflicted by the Amyloid Cascade: driving abnormal forms of the protein tau to spread beyond the brain neurons in the medial temporal lobe into the neocortex.
Fortunately, rejuvenation biotechnologies targeting aberrant tau in development, ranging from the earliest Petri dish studies all the way up to Phase III clinical trials. In part, this ambition to clear out tau started off as a hedge against the unfounded skepticism about the role of beta-amyloid in AD that set in during the years leading up to the breakthroughs with lecanemab and donanemab. One upside of this premature obituary-writing was that pharma companies and biotech startups started paying more attention to other AD targets, and aberrant tau was at the top of the list.
Although biotech companies were forced to abandon many of their first attempts to target tau (often because they interfered with the physiological function of the healthy, non-aggregated tau protein), pharma companies are currently running clinical trials to test numerous therapies targeting tau. Most of these potential therapies are in Phase II, meaning trials that are beyond basic safety testing, but won’t yet establish whether a given treatment actually bends the curve on AD or lead to approval by regulators like the FDA.
An important limitation of these candidate therapies is that most of them only clear tau aggregates located outside of cells — in the spaces where neurons interact with one another or in the wider fluid that bathes the brain. Such therapies should do some good, most notably by slowing down the rate at which “seeds” of aberrant tau “infect” other neurons in their network. But they would do nothing to clear existing aberrant tau in the location where those seeds form and where they inflict most of their harm: inside of neurons.
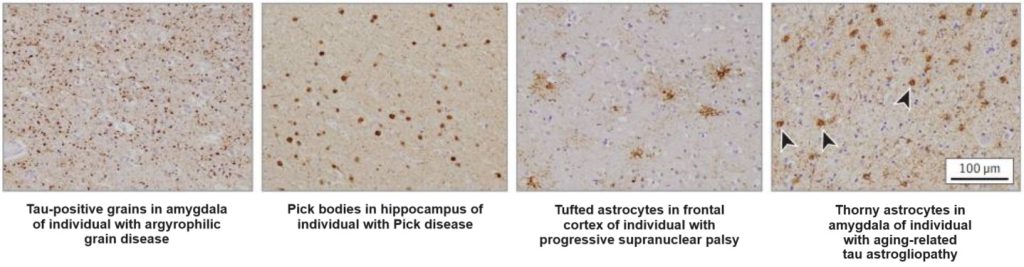
The main reason for this upside-down prioritization is that it’s not obvious how you would target aberrant tau inside neurons. We can’t use the original LysoSENS strategy to develop novel enzymes that would break down aberrant tau inside the lysosome, because tau aggregates don’t accumulate in the lysosome! Instead, they accumulate in the main body of the cell.
And we can’t turn around and deliver such enzymes into the main body of the cell either. First, they wouldn’t likely be effective: just as the stomach relies on stomach acid to help digestive enzymes break down protein in our food, one of the things that allows lysosomal enzymes to shred their targets so efficiently is that the lysosome maintains a very acidic internal pH, which would be incompatible with life if extended into the rest of the cell. And conversely, any enzyme so powerful as to destroy aberrant tau in the neutral pH of the cell body would likely be corrosive toward essential cellular proteins or interfere with essential biochemical reactions.
Most of the damage-repair therapies that are currently in clinical trials to target tau are instead AmyloSENS antibodies that work the same way donanemab and lecanemab do: by binding to their target in the fluid surrounding the brain and “pulling” it out into the circulation for eventual disposal. But (again) the bulk of the aberrant tau in the brain — and most of the damage it does — is inside our neurons. Antibodies don’t normally penetrate cells — and if they do, they are often sealed away in membranes that prevent them from doing much of anything once they get there.
And even for the small number of candidate tau-targeting antibodies that do seem to get into cells and escape the membraned chambers that bring them there, two factors make most of them the kind of exception that proves the rule. First, the number of such antibodies that manage to make it through the obstacle course we’ve just mapped out would be woefully too few to make a dent against the burden of aggregates each neuron contains, and would be spread too thin to get good coverage of all the brain cells that need them. And second, the main things that these particular antibodies could do are very inefficient, such as interfering with the glomming-together of individual tau molecules into aggregates or increasing their degradation (which would be a mixed benefit in humans with AD or other neurodegenerative aging diseases — more on this later).
And there is at least one case where an antibody that targeted soluble tau oligomers penetrated brain neurons and protected them against tau aggregate toxicity in mouse models, but lost this ability once it was tweaked for use in humans.
If we are to use antibodies to neutralize the threat to our minds posed by aberrant tau, we must at minimum find a way to deliver them into cells and keep them active once they get there. For this reason, Dr. Aubrey de Grey was excited several years ago when he noted a report that City of Hope scientists had developed a novel way to smuggle free antibodies into cells. At his instigation, SENS Research Foundation scientists worked for the last several years to use this tech to deliver antibodies against tau oligomers into cells — though after an initial roadblock, they are awaiting new funding to start again with the same core strategy from a clean slate.
Fortunately, other scientists are working on entirely new ways to get tau-targeting therapies into cells.
Onshoring Antibody Production
In recent years, scientists began targeting tau inside neurons using functional fragments of antibodies with key subunits that target different segments of the tau protein. When they treated animals that carry forms of mutated human tau that aggregate and drive neurodegenerative disease in humans, these antibody fragments lowered the burden of soluble aberrant tau in these animals’ brains — and the effect was almost entirely the result of clearing it from inside the neurons.
Following up, two groups of scientists pitted different tau-targeting antibody fragments against each other, and also against the same fragments after further modifying them to turn them into intrabodies. Intrabodies are antibody fragments that have been engineered to remain within and function inside cells, sometimes including targeting them to specific locations within the cell. When the researchers tested their tau-targeting antibodies and their intrabody equivalents in two different mouse models of tau-driven neurodegeneration, the tau-targeting intrabodies were more effective.
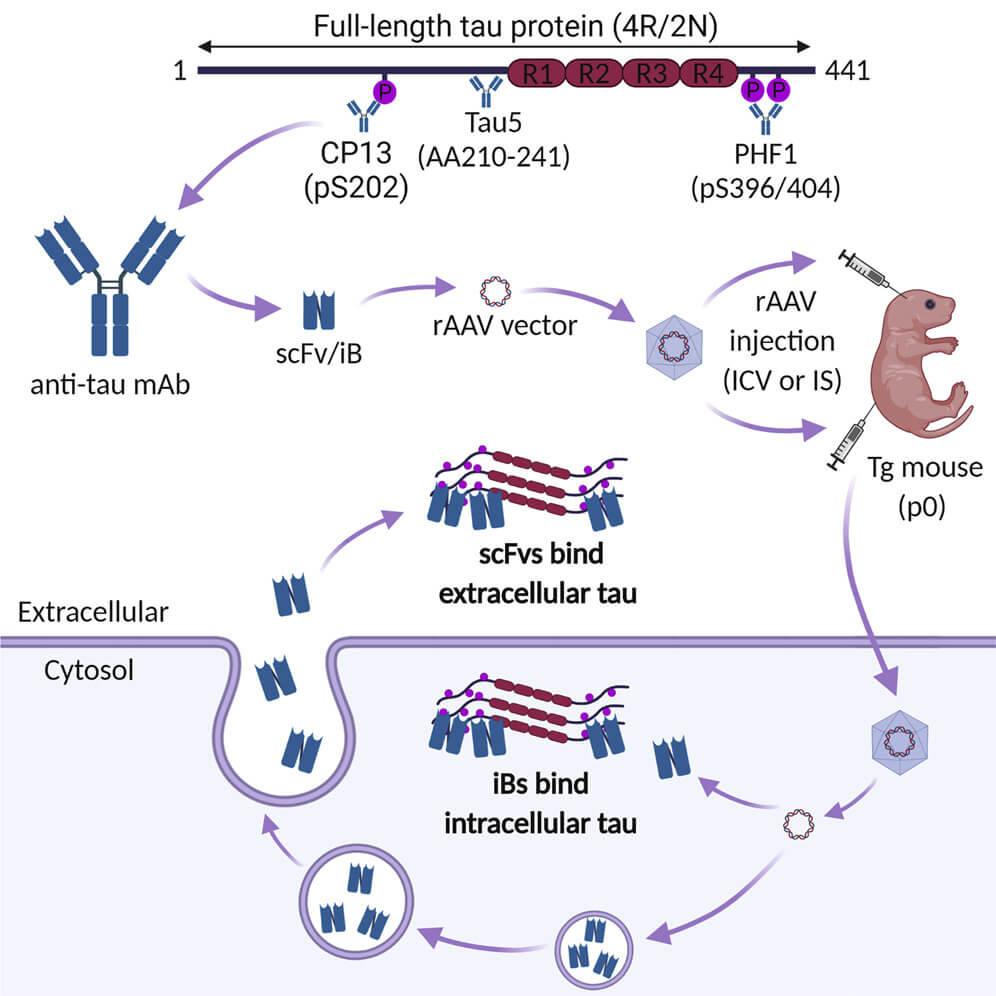
But how would we deliver these intrabodies to aging humans? All of these studies delivered the antibody fragments and intrabodies using gene therapy of some kind — either engineering the genes for these therapeutic antibodies into mouse embryos (which won’t work for those of us who, as Dr. Aubrey de Grey says, have “the misfortune of having already been born”), or deliver the gene in adult animals using specific strains of adeno-associated viruses (AAV) that target neurons. While AAVs are a leading option for gene therapy for many diseases, they have disadvantages we’ve discussed before, which will multiply the more we rely on them because of immune reactions to the AAVs themselves (although there are proposed ways to ameliorate that problem).
And true gene therapy has the possible disadvantage of being irreversible. If the safety and effectiveness of the therapeutic gene were well-established, it would be fine — even advantageous — to have a one-and-done round of gene therapy that would keep producing intrabodies targeting aberrant tau in one’s neurons indefinitely. But it would be hairy to run trials of such a therapy in people who (ideally) would still have a low burden of aberrant tau in their neurons and were minimally cognitively impaired. And turning the therapeutic gene off if something goes awry would be difficult without complex additional engineering of the therapeutic gene.
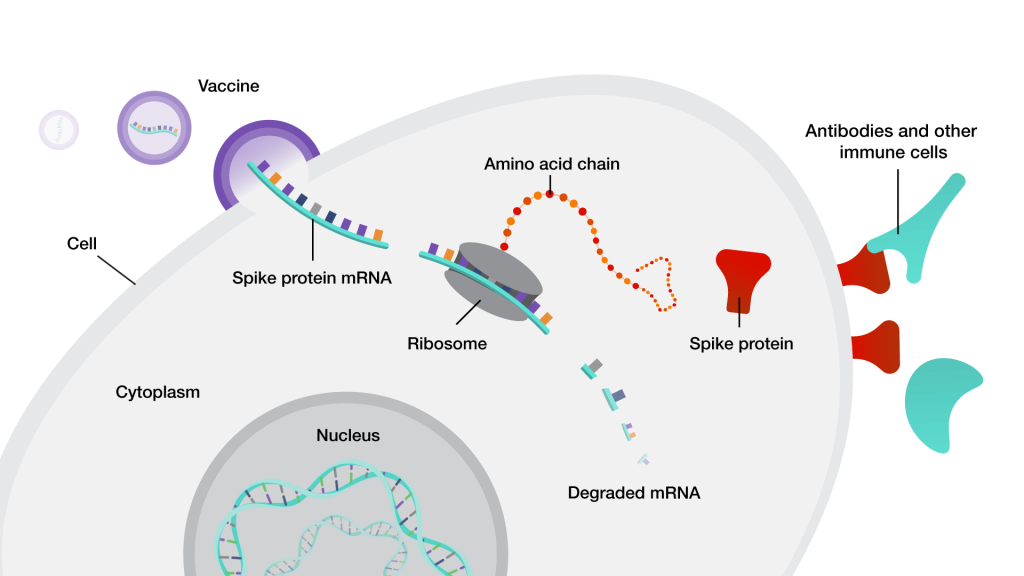
Researchers at the Florey Institute of Neuroscience and Mental Health in Australia have seized on the recent revolution of mRNA technology as a way to bypass these problems. Of course, we’ve all become familiar with mRNA as the platform for the leading COVID vaccines. mRNA is the “working copy” of a gene: when a cell wants to make a protein, it transcribes an mRNA version of the instructions for making that protein from the gene in the cell’s nucleus, and then the cell’s machinery follows the instructions in the mRNA to actually assemble the protein.
The COVID vaccines contained mRNA for a segment of the virus’ Spike protein, encased in special fat-based shells called lipid nanoparticles that keep the mRNA stable and deliver it into our cells. Once the mRNA gets inside the cell, the cell’s protein-production machinery gets to work producing the Spike protein — no virus or gene therapy required. And then the cell raises the Spike protein on its surface like a flag to the immune system, which sees that the protein is foreign and “learns” to be on the lookout for it should an actual COVID virus invade the body.
Combatting a global infectious pandemic was not the original purpose for which the pioneers of mRNA technology developed it. But now that it has proven itself for that purpose, scientists have seized on the “Warp Speed” advances in the technology to develop everything from personalized cancer vaccines to treatments for inherited genetic disorders. The Florey researchers realized that they could use mRNA to deliver the instructions for tau-targeting intrabodies into neurons. The cells would then produce free, active antibodies inside of themselves from scratch, avoiding the entire fraught journey required for conventional antibody infusions to reach and become active in the cell without requiring gene therapy.
The researchers started off by engineering mRNAs based on a conventional tau-targeting antibody called RNJ1 that their team lead had developed previously. In this new study, they confirmed that RNJ1 was able to bind tau aggregates derived from human brain tissue, but its effects were underwhelming in a previous study in which the researchers tested it in mice bearing a human mutation in tau that leads to neurodegenerative disease. In that study, infusing the conventional RNJ1 antibody into the mice lowered their burden of abnormal tau and improved the function of their neurons, but it didn’t improve their abnormal motion control. Could an intrabody version of RNJ1 do better?
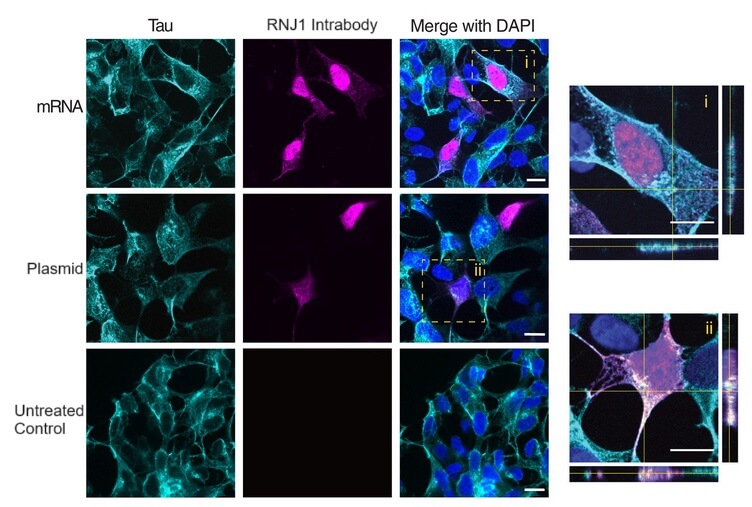
As a first step toward finding out, the Forey team produced two mRNAs: one that would instruct cells to produce the conventional RNJ1 antibody and secrete it, and another with instructions for an intrabody version of it. When they delivered each mRNA into human neuron-derived cells, each mRNA triggered the cells to produce its respective antibody. The conventional RNJ1 antibody was produced from its mRNA in two halves, which assembled into the final antibody and was secreted outside the cell as they had intended. There, it bound to tau in the fluid bathing the cell.
Meanwhile, the intrabody version of RNJ1 remained within the neuron-like cells. Promisingly, the cells produced more of the RNJ1 intrabody from mRNA than they did when the same intrabody was encoded in a plasmid, which is a more conventional lab technique for producing proteins in the main body of the cell.
The Florey team then delivered the intrabody mRNA or the plasmid to human neuron-like cells that also express a tagged form of human tau. Both intrabody delivery systems produced intrabodies that appeared in locations inside the cell where tau was also located, suggesting they were homing to tau and binding on. The reasearchers then did an additional assay to confirm that the intrabodies and plasmids were actually interacting with tau.
Early Days for a Long Brain Life
This research is promising, but is still in a very preliminary stage. The Florey team previously tested RNJ1 in a mouse model but hasn’t yet tried out the intrabody version beyond these initial cell culture studies. While the potential of an mRNA vaccine against aberrant tau is exciting, several hurdles remain to be overcome.
The first of these is access to the brain. As we noted earlier, scientists have to package therapeutic mRNA up into protective lipid nanoparticles to keep it from being degraded as it travels the body. However, the brain is in a kind of biological Fortress of Solitude, protected against foreign materials by a layer of cells that are tightly wedged together in the lining of the blood vessels that feed it. As a result, no one has yet figured out how to get lipid nanoparticles into the brain after a simple injection. However, there’s a lot of excitement about using mRNA to target cancers and other health problems in the brain, so hopefully someone will develop a working delivery system soon — perhaps by changing the composition of fats in the nanoparticles, or by developing a way to let them port in via one of the many transport systems that allow essential biological molecules to get inside the cerebral bunker.
Another area where more work is needed is the underlying antibody from which the intrabody is made. To be frank, RNJ1 is not a particularly promising place to start. As we noted earlier, RNJ1’s effects in a mouse model were quite limited, both in terms of how much it lowered abnormal tau and in terms of improving the function of the mice. Part of the reason may be that it grabs onto the same region inside the tau protein as did three other antibodies that failed in human clinical trials.
But a more important reason, also shared by most of those antibodies, is that it goes after tau in an indiscriminate way: it not only binds to some kinds of abnormal tau, but also to normal, undamaged tau protein. This may actually be part of how it works: by binding up normal, unaggregated tau, less tau is available for the “seeds” of aggregated tau to twist into new aggregates, which then reduces the number of new tau aggregates that get seeded in the brain.
That would explain the results of a recent mouse study in which scientists treated mice that harbor a human tau mutation with a conventional antibody that exclusively targets normal tau. Mice that received the antibody went on to suffer fewer tau aggregates and lost fewer neurons going forward than the control mice. Yet, the antibody didn’t protect them against the degenerative motion disorder that the mutation causes.
Developing a true damage-repair intrabody that targeted aberrant tau exclusively and removed pre-existing aggregates would be a much more powerful strategy. It would also reduce the risk of side-effects related to robbing the cell of normal tau, which the cell needs as a scaffolding protein that also helps shuttle proteins along its length like a monorail. There are several strong conventional antibody AmyloSENS candidates that target aberrant tau and might be tweaked into intrabodies instead, such as tau oligomer-specific monoclonal antibody (TOMA) (which targets tau oligomers) and two antibodies that target ptau-217, a specific form of abnormal tau that looks promising as both a biomarker for tau-based molecular damage and also as a target for damage-repair longevity therapeutics. One of these antibodies, Janssen’s JNJ-63733657, is currently in a Phase III human clinical trial, and another recently showed promising results in a mouse model.
And having bound to aberrant tau inside the neuron — well, what then? As we noted above, in the few cases where regular tau-targeting antibodies do get inside neurons and manage to break out of their entry bubbles and bind to aberrant tau in the neuron, their ability to clear out tau is limited, and it’s not entirely clear how they are able to do what they do. Likely, most of them keep tau from assembling into aggregates, or even break early aggregates up, and some of them may help the cell’s protein-degrading machinery tear them apart.
A more powerful approach, which SENS Research Foundation scientists have worked to develop with the novel delivery system we mentioned earlier, is catalytic antibodies (catabodies) to chop pathological aggregates into harmless debris that the cell could then mop up on its own. Our scientists were endeavoring to shuttle tau-targeting catabodies into the cell, but this study suggests that promising catabodies could instead be encoded in mRNA and produced on-site inside the neuron.
The good news, again, is that there is enormous enthusiasm amongst researchers and biotech companies in harnessing mRNA as a delivery system to deliver therapies for a wide range of unrelated diseases, from congenital genetic disorders to cancer. Because mRNA is a platform for delivering therapies rather than a disease-specific or target-specific therapeutic approach, solutions for the challenges that face scientists working to use mRNA for one use case are likely to quickly spill over into other uses, including tau-targeting intrabodies or catabodies. In fact, this pilot study in using mRNA to deliver intrabodies targeting tau is not even the first case of mRNA being used to deliver a longevity therapeutic: we are aware of a biotech company working to use this platform to deliver a LysoSENS therapy targeting a completely unrelated intracellular aggregate on damage-repair principles, and other longevity biotech companies are likely to come up with good use-cases for targeting other forms of cellular and molecular aging damage.
We’ve already seen evidence that people enjoy greater benefits from AmyloSENS therapies like lecanemab and donanemab when their burden of aberrant tau is lower rather than higher. The clear implication is that they would get even more benefit if their existing burden of aberrant tau were cleaned out — perhaps before receiving amyloid-clearing therapies, perhaps after starting on them. Using mRNA to deliver antibodies designed for damaged proteins inside cells is another key piece in the puzzle of keeping our minds clear and sharp and holding onto our memories and identities into an indefinite future.
