

SENSible Question: Everyone knows that mitochondrial free radicals are a key driver of aging, and antioxidants don’t seem to offer any protection. Birds are supposed to have very clean-burning mitochondria, so should you maybe try to cut them off at the source by re-engineering our mitochondria to be more like those of birds?
It’s definitely a puzzle. Our bodies’ normal metabolic processes continuously generate free radicals, which are highly reactive molecules with at least one unpaired electron in their outer shells (such as reactive oxygen and nitrogen species) or that act as if they were (such as some transition metals). On top of that, environmental stressors constantly bombard our bodies with even more free radicals: everything from ultraviolet light, to subtly rancid fats, to some metals and air pollutants.
Because free radicals are highly reactive, they frequently damage the working components of our bodies. And while our metabolisms quickly repair some of these damaged structures and break down others for reuse or excretion, occasionally these radicals irreparably damage long-lived structures in our bodies, making them dysfunctional for the long term — or even the rest of our lives.
And beyond free radicals’ damaging effects on the molecules and organelles that keep us alive and healthy, the burden of free radicals gradually gets the upper hand over antioxidant molecules and other control systems in our bodies, leading to a chronic imbalance (oxidative stress) with age. Oxidative stress not only further increases the amount of free radical damage we suffer each day, but also deranges normal signaling pathways in the body, feeding cells molecular misinformation that makes them behave like your conspiracy theory-obsessed uncle at Thanksgiving.
All of that’s just got to be bad for a person! And certainly (on the one hand) bombarding experimental animals with different sources of free radicals shortens their lives, while (on the other hand) cells from longer-lived species or from mice or other experimental animals that receive anti-aging interventions are generally more resistant to oxidative stress. So it seems obvious that reducing an organism’s exposure to free radicals would lengthen its life.
And yet, scientists have tried again and again to mitigate those harms using antioxidants and have never nudged mammals to live longer, with one important exception that we’ll briefly discuss later on. Whether they tried feeding antioxidant compounds, or upregulating animals’ internal antioxidant enzymes, or even in some cases deleting the genes for those enzymes entirely, nothing seems to move the needle in mammals (though it often does in invertebrate models like worms and flies).
Meanwhile, clinical trials of antioxidant supplements to delay or prevent cancer and cardiovascular disease in humans have consistently been failures at best, and at worst have even increased the death rate among users. High-dose (200 micrograms) selenium, specifically, also pushed many trial participants into diabetes or impaired glucose tolerance in two large clinical trials (the Nutritional Prevention of Cancer Study and the even larger SELECT study); that’s consistent with what epidemiological studies have found when they have looked at people in the United States, where background selenium intake is already high. And not only do antioxidant vitamins C and E not slow down the rate of cognitive decline in healthy people, a small trial of C and E plus lipoic acid found a signal that antioxidant treatment accelerates cognitive decline in people with neurodegenerative aging of the Alzheimer’s type.
Much of this lack of effect is thought to be because antioxidants aren’t very effective at actually lowering oxidative damage, or at lowering it in the places and under the conditions where it causes irreversible harm. We’ll get back to this idea later on in this post. But there’s a more profound reason why preemptively setting your molecular fire sprinklers to run all the time is not a good solution to the problem of free radical firebrands.
As we alluded above, the body needs free radicals to carry out many functions, including harnessing them as signaling molecules. Evolution has built molecular “triggers” into regulatory proteins that are intentionally vulnerable to damage from free radicals. When the “trigger” is oxidized, it twists the protein’s shape in a way that turns it into a “key” that “unlocks” other proteins that allow the cell to adapt to the rise in stress. Different cells and systems even generate free radicals themselves to pass on signals and carry out important biochemical functions. Among other things, free radicals transmit biochemical messages that tell the cell to grow, reproduce itself, move in particular ways, or commit “cellular suicide” (apoptosis) to protect the body as a whole from the cell’s own threat to it.
We covered two of the more recently discovered examples of this in a previous post. First, fasting and other stimuli activate autophagy by generating free radicals that oxidize Atg4, one of the proteins involved in regulating it. In one experiment, antioxidants blocked rapamycin’s ability to stimulate autophagy and thereby degrade damaged proteins involved in neurodegenerative disease.
Second, oxidative stress allows intestinal stem cells to begin proliferating. Most of the time, these stem cells are kept dormant because a protein called Nrf2 tamps down oxidative stress in the cell by turning on the genes for multiple cellular antioxidants. By preventing intestinal stem cells from proliferating continuously, Nrf2 preserves the tissue’s stem cell reserve for when the tissue is damaged and replacement cells are needed, which we might imagine might be accompanied by a burst of oxidative stress. But if that burst of free radicals were blocked, the cells would remain asleep at the wheel and the tissue would not be repaired.
A third example with important practical implications is the role of free radicals in adaptations to exercise. When you work your muscles, it generates a burst of superoxide radicals and nitric oxide that triggers the tissue to adapt to the challenge you’re putting it through. One set of responses helps your muscles adapt to the free radicals themselves by producing more antioxidant and other protective factors to shield the muscle from damage, but others are the kinds of health and fitness adaptations for which we do exercise in the first place.
So what happens when you suppress these free radicals with antioxidants? Double-blind trials in humans have found that antioxidant supplements block the blood-pressure-lowering and blood vessel responsiveness benefits of exercise, as well as the ability of exercise to blunt fat tissue hormones that contribute to insulin resistance. Moreover, a rodent study found that exercised rats don’t produce as many new mitochondria or surge their mitochondrial activity as much if scientists supplement their chow with antioxidants.
And beyond using free radicals as signaling molecules, the body also puts them to use in more direct ways, such as when immune cells generate free radicals to destroy pathogens. (A paper from 2010 that got a lot of press coverage for its claim to show otherwise was subsequently retracted). The body also uses free radical chemistry to make, modify, and shape many of the body’s proteins and to detoxify drugs and other foreign molecules.
So when you flood your system with antioxidant molecules, one inevitable downside is that you’re blocking free radicals that the body needs to carry on the business of life. Those same radicals could also slam mindlessly into cell membranes, damaging their function, but there’s no way to exclusively squelch radicals that damage important biological structures without also sweeping up radicals that the body needs for signaling purposes.
A crude version of the idea of engineering our mitochondria to be cleaner-burning is to force the cell to produce fewer free radicals by interfering in different ways with the activity of mitochondria, which are both a major source of free radicals in the cell and a critical target for free radical damage that limits lifespan. Some scientists have argued that the diabetes drug metformin might be one way to achieve this.
Metformin does a lot of things in the cell, and the main molecular mechanism responsible for its blood sugar-lowering effects is hotly contested, but one potential mechanism is that it blocks the shuttling of electrons into Complex I of the mitochondria. Because Complex I is one of the two main sites from which electrons “leak” out of the mitochondria during energy generation, some researchers have argued that inhibiting this Complex with metformin might lower free radical production in the process. Today, many scientists doubt that metformin inhibits mitochondrial function in living metformin users (and not just cells or tissue culture); this would explain why metformin very clearly does not increase lifespan in mice or in nondiabetic people (see also here).
A totally different shot on the goal of reducing mitochondrial free radical production is to “drain off” some of the electrochemical “reservoir” that drives mitochondrial energy production. Once some of the mitochondria’s potential energy has been dissipated as heat instead of being maintained to drive cellular energy production, you’d expect the mitochondria to generate fewer free radical byproducts of the energy-producing machinery and thus inflict less damage on themselves, leading to extended lifespan.
The drug dinitrophenol (DNP) certainly does the first half of that job, raising people’s metabolic rate by a remarkable 40%, resulting in a massive increase in body temperature and a drawdown of multiple cellular energy reserves. The scientists who first reported these effects noted DNP’s potential as a weight loss drug, but cautioned about the toxicity that had been reported in animal studies. Heedless of their warnings, 20 companies sprung up within a year of their report to sell DNP and related molecules as weight-loss drugs. Many people self-medicating with DNP for weight loss died feverish deaths in the 1930s until the law was updated in 1938 that allowed FDA to step in. When FDA announced that it would prosecute any sales of DNP through interstate commerce, the DNP pill mills largely pulled up stakes.
Despite this record of toxicity and death (extending right up to the present day), scientists reported in the early 2000s that dinitrophenol actually extended the lifespan of mice, as you might expect if it had indeed short-circuited free radical production in the mice’s mitochondria.
The problem with this claim is that the scientists used a short-lived strain of mice in the experiment, so the “increased lifespan” reported in the DNP-treated animals was nothing more than restoring something closer to the normal lifespan of an otherwise-healthy aging mouse to the treated animals. And indeed, when tested in healthy mice by the NIA’s Interventions Testing Program (ITP), DNP had no effect on lifespan at all.
This problem of would-be longevity therapeutics looking good only because they improve the deranged metabolism of sickly mice is a chronic flaw in rodent lifespan studies. Steven Spindler and many others have been raising the alarm about this for decades, and biogerontologist Kamil Pabis and distinguished colleagues are trying to set a “900 day” scientific standard to purge the field of such “extended (normal) lifespan” claims. In the meantime, be on guard for this kind of “longevity inflation.”
Build a Better Mouse?
The inherent problems with either indiscriminately quenching free radicals or putting a throttle governor on energy production in the mitochondria help explain why evolution makes so little use of antioxidants when conditions in an animal’s niche create selective pressure to lengthen a species’ lifespan. Surprisingly, in fact, the overall pattern is for longer-lived species to have lower expression of antioxidant enzymes, as SRF founding CSO Dr. Aubrey de Grey pointed out some years ago. More recently, this trend was extended in studies comparing the antioxidant expression in shorter-lived mice versus long-lived naked mole rats (NMR).
Instead of trying to capture free radicals before they can tear into an animal’s long-lived structures, evolution builds animals to be longer lived by making their metabolism “cleaner” (less prone to making free radicals during mitochondrial energy production and other metabolic activities) and their structures more able to stand up to free radical damage. Think of it like designing an engine to last longer. On the one hand, you would build it out of higher-quality parts that can withstand the effects of friction, mechanical stress, and sludgy oil. And it will last even longer if you also engineer it to crank more smoothly and produce less waste heat so it doesn’t degrade its oil so much in the first place.
Among the ways genes can harden an animal’s long-lived structures against free radical attack are using fewer easily-oxidized sulfur-containing amino acids when building long-lived proteins (including possibly replacing the amino acid cysteine with a less-oxidizable amino acid in sensitive regions of the mitochondria) and using unsaturated fatty acids with fewer highly-oxidizable double bonds when building cellular and (especially) mitochondrial membranes. Evolution can also select for variants of repair proteins that patch up the early lesions in biological structures more aggressively, thereby preventing them from progressing to become irreversible.
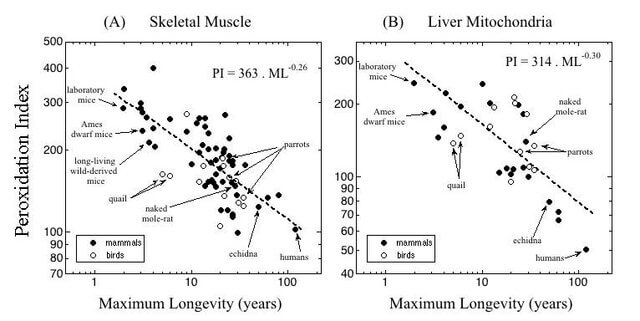
As an example, let’s again consider the homely but long-lived NMR. It’s often been said that NMR survive much longer than their mouse and rat cousins despite bearing an enormous amount of free radical damage in their tissues. But this only tells part of the story. First, the “oxidative damage” that people refer to when they say this is not “damage” in the SENS sense of the term (stable, structural defects inflicted on cells and long-lived biological structures that accumulate over time in one’s tissues). Instead, this “damage” is the initial “hits” by free radicals onto biological molecules that can eventually mature into such damage, but that the body still has the opportunity to repair, degrade, or excrete.
For example, NMR live with a high steady-state level of 8-oxo-2′deoxyguanosine (8-OHdG — an early lesion caused by the attack of free radicals on the DNA) in most of their tissues (though they have a quite low level in their livers and kidneys). But the animal’s DNA repair machinery is capable of removing 8-OHdG before it results in an actual mutation — that is, the kind of irreversible change to the genome that counts as real “damage” in the SENS sense.
One way that NMR keep these early insults from progressing into permanent damage appears to be that they have highly active repair and degradation machinery compared to shorter-lived species (such their DNA repair enzymes and the protein-degrading machines in their cells). Beyond that, in some cases NMR genes “design” their biological structures to be more resilient in the face of careening free radicals than mice do. These sorts of factors explain how NMR can go about their business with higher steady-state levels of early oxidative damage to lipids and oxidatively-damaged sulfur-containing amino acids in their proteins than shorter-lived mice, and yet escape the progressive rise in the level of these early insults in the back half of their lifespans that mice and other studied species (including humans) suffer.
For example, NMR build their membranes to be more resistant to free radical damage as compared with their shorter-lived rodent cousins. As you can see from the graph above, they pack far fewer of the most oxidation-prone unsaturated fatty acids into their cellular membranes than shorter-lived mice do — though not astonishingly so: they are in line in this respect with other animals relative to their lifespans. And they maintain the youthful makeup of their membranes throughout life, whereas many animals begin to let more oxidizable fatty acids creep into their membranes with age. Slow-aging Ames dwarf mice and mice on anti-aging Calorie restriction also shift their metabolism to keep fewer highly-oxidizable fatty acids in their membranes.
Moreover, NMR’s proteins are exceptionally resistant to types of damage not driven by free radicals, which perhaps makes it easier for them to carry on in the face of their high burden of specifically oxidative damage.
Turbocharge the Mitochondria?
Turbochargers are best known as a technology for making cars accelerate more quickly, but for the last two decades they’ve been quietly put under the hood of most cars and trucks so that they can deliver more power with fewer emissions. In the case of the evolution of longer lifespans, the analogy is to constructing mitochondria and other biological machines in a way that allows them to generate the same amount of cellular energy with fewer free radicals. Specifically, the working parts of longer-lived animals’ mitochondria handle electrons more tightly, so that fewer electrons careen off their intended course and form free radicals. By preventing the unintentional production of free radical waste, these animals’ mitochondria suffer less oxidative damage, but can still take advantage of free radical signaling from intentional self-generated sources and events in a changing environment.
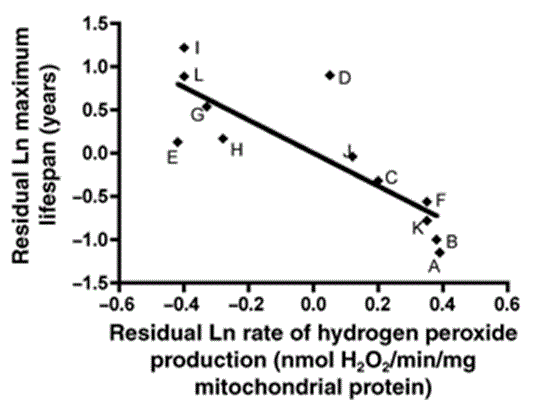
Most importantly, tight-running mitochondria are less prone to damaging themselves. The specific importance of mitochondria as a source and target of free radical damage is evident from things like:
- the fact that oxidative “damage” to mitochondrial, but not nuclear DNA is correlated to species maximum lifespan (see the figures above and below);
- that this dichotomy is true even though nuclear mutations (which can be caused by many kinds of damages other than free radicals) are strongly correlated with lifespan;
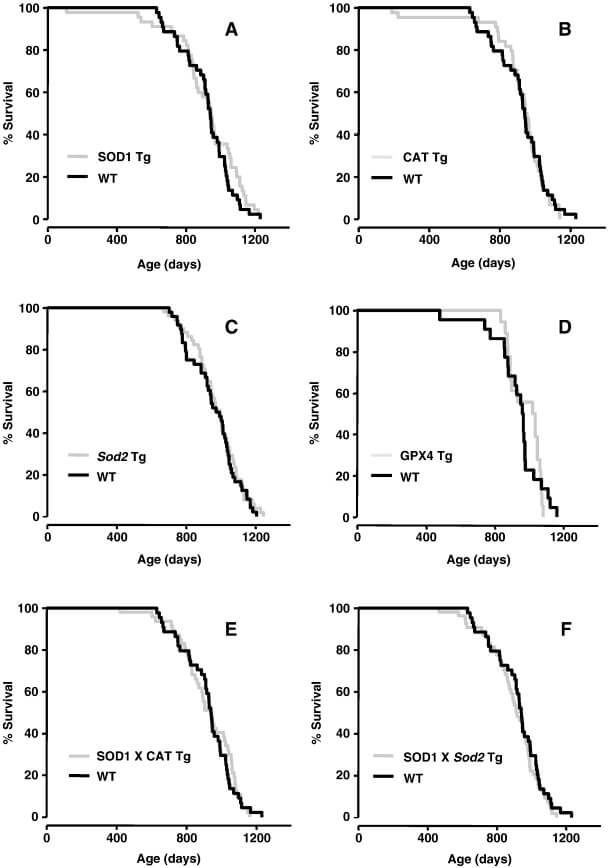
- that targeting the antioxidant enzyme catalase to the mitochondria extends lifespan in mice, whereas (as we mentioned earlier) no antioxidant enzyme expressed elsewhere in the body does so, including catalase in its normal location in the main body of the cell (see the first figure in this blog post);
- that two major slow-aging interventions (Calorie restriction and the Ames dwarf mutation) cause changes in mitochondrial metabolism to produce fewer free radicals and suffer fewer mitochondrial deletion mutations; and
- the fact that so many of the toughest diseases and disabilities of aging are tightly linked to mitochondrial DNA deletions, such as Alzheimer’s and Parkinson’s diseases and sarcopenia (the aging disease that causes even lifelong athletes to not only lose muscle mass as they age, but to lose strength to a degree that is proportionately even greater than the amount of muscle lost).
Fix Your Wheel — Don’t Reinvent It
All of this background leads into our question for this blog post. When people hear about the much lower rates of free radical production in the mitochondria of birds or the oxidation-resistant structures of NMR compared with similar-sized animals, they often seize upon the idea that we should engineer those same features into our own cells to slow the rate of aging in humans. This isn’t a sound strategy for four reasons.
First, although articles often emphasize how much more efficient the mitochondria of longer-lived birds are relative to the mitochondria of shorter-lived mice, there’s no reason to think that pigeon mitochondria are any cleaner-burning than human mitochondria already are. You can already see from the graph above that birds are not anomalous in this way: longer-lived animals consistently generate fewer free radicals in their mitochondria than shorter-lived ones do, especially when adjusted for body size. And the logical prediction from that trend is that one of the reasons humans can live to be greater than 85 years old while pigeons live no more than 35 years is that we already have much lower mitochondrial free radical production than they do.
Unfortunately, we don’t have a single study that uses the same methods to compare free radical production by the mitochondria of longer-lived birds versus those of humans to prove this point. But we can get very close by “triangulating” the data in the graph above with those in a more recent study that used modern methods to compare the maximum lifespans and mitochondrial free radical production rates among primate species, with mice added for reference. As you can see in the graph below, the same inverse correlation between maximum lifespan and mitochondrial free radical production holds among primates, with human cells’ mitochondria producing far fewer free radicals under standard conditions than do those of any of our shorter-lived primate cousins, let alone mice. In fact, we have even cleaner-burning mitochondria than you would predict from the trend line among primate species!
Now note in particular the comparison between us and baboons (P. hamadryas), which are somewhat below the trend line for their lifespan. And while we can’t draw the following conclusion with certainty, because we’re comparing different studies using different methods to measure mitochondrial free radical production, you can see in the previous figure from Aging Cell (above) that baboons are also slightly below the trend line for lifespan vs. mito free radical production across a range of other species with widely-varying lifespans — and that they are roughly in line pigeons, which have a similar maximum lifespan (37.5 and 35 years, respectively).
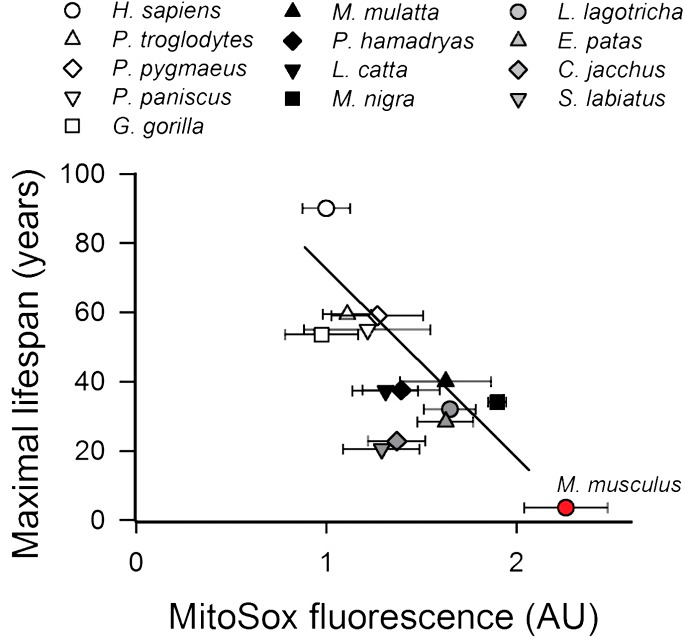
So contrary to the assumption behind the question, we can say with a high level of confidence that swapping out human mitochondria for those of birds would actually leave us with dirtier-burning cellular power plants, not high-efficiency models.
To explain the second problem with the proposal to “avianize” our mitochondria, let us suppose for the sake of argument that birds’ mitochondria really did produce less free radical waste than their human equivalent — like moving from mouse mitos to pigeon power plants. To get our mitochondria to work as efficiently as theirs do, we would have to completely rejigger most or all of the genes that encode for the different working parts of the mitochondria’s energy-production drivetrain, and potentially also the genes for other cellular and mitochondrial machinery that interacts with them. It would be like trying to put together a jigsaw puzzle in the dark, knowing from memory approximately what the final picture should look like but not being able to see what’s on each piece you’re trying to fit together.
And we would not only have to ensure that the final powertrain line would continue to generate adequate cellular energy while producing substantially fewer free radicals, but also that they could still carry out all the other functions that mitochondria perform for us. This includes detoxifying ammonia, raising our body temperature (birds are warm-blooded, but they have feathers for insulation and much higher body surface-to-volume ratios to let off heat), responding to signals for programmed cell death, balancing the cell’s oxidation-reduction chemistry, and synthesizing products that include amino acids like taurine, heme (the protein that carries oxygen in our red blood cells), steroid hormones, and the “letters” that encode our DNA.
And supposing we could somehow figure out a way to do the modeling and iterative experimentation to design and synthesize all the genes that would be needed to make such mitochondria (understand: I’m driven to despair by the magnitude of this project!), how would we turn it into a working rejuvenation biotechnology?
Compare it to what Dr. Boominathan and her colleagues at the SENS Research Foundation MitoSENS lab are already charged with doing: placing “backup copies” of the 13 mitochondrially-encoded protein-coding genes in the nucleus. As it stands, “mere” allotopic expression of these 13 genes is already challenging, following which we will need to deliver these engineered mitochondrial genes in vivo in mice and then in humans who have already been born. But in this project, “all” the engineering the team is doing is to make nuclear versions of our existing mitochondrially-encoded genes, without giving them any new properties. “Avianizing” our mitochondria would entail re-engineering not just the 13 protein-coding genes that are encoded by our mitochondria themselves (as in our existing allotopic expression project), but all 102 genes in the mitochondria and the nucleus combined that code for different parts of the mitochondrial powertrain.
And on top of that, you would also expect to have to make tweaks or wholesale overhauls of many of the 1,100-1,500 other mitochondrial genes encoded in the nucleus that code for parts of the mitochondria other than the energy-producing drivetrain itself so that they would properly interact with it.
Because we don’t have an effective way to do true mitochondrial gene therapy, we would still have to perform allotopic expression for the 13 “avianized” mitochondrially-encoded proteins, and now add on top of that more conventional gene therapy for the avianized versions of those that are naturally encoded in the nucleus. And it’s even harder than the sheer numbers imply, because unlike when we engineer “backup copies” of our original mitochondrial genes into the nucleus, we can’t allow our original human mitochondrial genes to keep working alongside the avianized versions. For this “avianized mitochondria” project to succeed, we would have to disable all of our original human mitochondrial subunits to keep their proteins from interfering with the assembly of the avianized version — like putting Honda replacement parts into a Chevy engine. Half-bird, half-human hybrids make for entertaining fantasy novels, but will not work for the purpose of creating ultra-clean-burning mitochondria.
On top of that, it’s recently been discovered that birds have mitochondria in their red blood cells (RBC), which adult mammals do not. No one knows exactly why birds hold onto these RBC mitochondria while mammals lose them during development, but circulating mitochondria might take some of the load off of muscle cell mitochondria for things like synthesizing heme for oxygen transport (which is the RBCs’ main job) or detoxifying substances in the circulation. These circulating mitochondria might also help birds cope with their diabetic-like high blood sugar levels. It’s not clear, therefore, whether “merely” swapping out the mitochondria in our solid organ cells with “avianized” versions would allow us to get by, absent also engineering our red blood cells to start making mitochondria again — and to make avianized mitochondria instead of the ones for which these cells still carry the genes but deactivated them in our first weeks of development.

The third reason why “avianizing” our mitochondria to produce fewer free radicals is not as strong a strategy to combat degenerative aging as it sounds is that it assumes an outsized role for the direct effect of mitochondrial free radicals on free radical damage elsewhere in our tissues under normal operating conditions (that is, before the mitochondria suffer major mutations with age). It’s long been thought (and I’ve often said) that mitochondria are the single greatest source of free radicals in the cell. But this belief turns out to rest on some questionable assumptions, and more detailed studies with more advanced techniques seem to refute the idea. Instead, although they are a substantial source, mitochondria are not the largest source of free radicals in the cell. That, plus the distance between the nucleus and the mitochondria relative to other sources of free radicals in the cell, explains why the impact of mitochondrial free radicals on the nuclear DNA is negligible.
Additionally, people often still have some version of the “vicious cycle” theory of mitochondria’s role in aging in their heads, or what we might call the “grandpa’s Lincoln Continental” theory. The “vicious cycle” theory supposes that a mitochondrion’s free radicals damage the mitochondrion itself, which causes it to become dysfunctional and generate more free radicals, thereby causing even more damage to itself, and so on and so forth until the cell looks like an ill-maintained 1970s boat of an automobile. But it’s been known for decades that the “vicious cycle” theory of mitochondria in aging is incorrect. Indeed, the signature mutation in long-lived aging cells is the so-called “common deletion,” which shuts down mitochondrial free radical production entirely because it disables the machinery of mitochondrial energy generation, which is the source of mitochondrial free radicals in the first place.
In fact, an important impetus for SRF Founding CSO Dr. Aubrey de Grey developing his quite baroque mitochondrial free radical theory of aging was the search for a coherent way to explain how mitochondrial free radical generation drives aging (as it clearly does) if the “vicious cycle theory” was wrong. In Dr. de Grey’s theory, mitochondria cause the most problems to the aging body after the cell is taken over by mitochondria that no longer generate free radicals at all. The systemic harm comes instead from the metabolic hoops that the cell has to jump through to keep a minimum level of energy production going once the mitochondria’s energy production drivetrain is offline.
It’s certainly true that having mitochondria that generated fewer free radicals in the first place would reduce the rate at which mitochondria inflicted the “common deletion” on themselves, which would in turn delay the conversion of their host cells into centers of oxidative stress for the rest of the body. But it wouldn’t cause the lifelong reduction in damage inside each cell that one would assume if one imagined a simple story in which the mitochondria were directly riddling other organelles and structures inside the cell with free radical damage.
The last problem with the idea of “avianizing” our mitochondria is the most fundamental. While going so far as to completely overhaul the entire apparatus at the heart of most cellular energy is a formidable, almost science-fiction bioengineering project, in one sense it’s a very old-school approach to the problem of free radical damage. Like statins, exercise, rapamycin, and DNP, it’s yet another attempt to force our metabolism to generate less aging damage, rather than a strategy to remove and repair existing damage in our tissues. Generating fewer mitochondrial free radicals would slow the rate at which our mitochondria pummel themselves into dysfunction, but it would do nothing to restore function in mitochondria that have already suffered large deletion mutations and rejuvenate our cells. However technically ambitious, it remains strategically complacent.
By contrast, the “damage-repair” strategies pursued by SENS Research Foundation will not deal with free radical damage by trying to prevent free radicals from being generated or catch them all like a frenetic Pokémon game. We will allow metabolism to do its thing, accepting that it will produce some free radicals. Some of them will act as useful signaling factors, while others will damage our mitochondria and other working parts of our bodies — and still others will do some of both. But then we will repair the damage before it accumulates to the point of causing disease or debility.
All the MitoSENS strategies under pursuit by the SENS Research Foundation MitoSENS lab — allotopic expression (“backup copies”), the “gene drive,” and a potential mitophagy “unmasking” drug — aim to repair or replace damaged mitochondrial genes, organelles, or proteins, thus making sure that our mitochondria keep working just as they do when we’re young, indefinitely.
So long as we can keep delivering functional components to the mitochondria to keep them working (allotopic expression), or clear out damaged mitochondrial genomes and replace them with pristine ones (gene drive), or restore the ability of the mitophagy machinery to “see” deletion-bearing mitochondria and cull them (“unmasking”), it doesn’t matter how quickly mitochondria damage themselves, except in the sense that it will dictate the frequency at which we deliver rounds of MitoSENS therapies.
And the same principles apply to other targets of free radical damage. If we clear lipofuscin out of our cells, we won’t have to worry about the contribution of free radicals to producing lipofuscin. If we destroy senescent cells on an appropriate schedule, we won’t have to worry about the contribution of free radicals to some forms of senescence. And so on.