
Lewy bodies (LB) and other intracellular α-synuclein (AS) aggregates accumulate in the aging nervous system, and a high burden of such aggregates are hallmark neuropathological signs of Parkinson’s disease (PD), Lewy body dementia (LBD), multiple systems atrophy (MSA), and other synucleinopathies. Loss of dopaminergic (DA) neurons in the substantia nigra (SN) to aging processes and toxicity are chiefly responsible for the most overt motor symptoms of PD (rigidity, bradykinesia, and resting tremor), and it is on the basis of these symptoms that PD is clinically diagnosed. But prior to the onset of motor symptoms, AS pathology is already present in the peripheral (particularly enteric) nervous system of aging and especially future PD subjects. This pathology is associated with a constellation of nonspecific and sometimes minor-seeming preclinical symptoms, including autonomic dysfunction (particularly constipation), olfactory dysfunction, psychiatric disorders including depression, and REM sleep behavior disorder, and the presence of such symptoms predicts future PD.[3] By the time PD reaches the clinical phase, AS pathology has progressed into the brainstem and caudally onward through the substantia nigra. This advance, and its future extension to the neocortex, is thought to be responsible for the most troubling disabilities associated with advanced PD, such as gait freezing, dysphagia, dysarthria, and worsening and non-levodopa-responsive postural instability and falling.[1,2] Cerebral AS pathology is also closely associated with the visual hallucinations that typify LBD, and it is becoming increasingly clear that LB along with other neuronal protein aggregates are key drivers of “normal” cognitive aging.
In a previous post, we surveyed an exciting new development in rejuvenation biotechnology: the sudden emergence and rapid progress toward the clinic of vaccine- and antibody (Ab)-based immunotherapy to remove α-synuclein aggregates from the aging brain. Therapies applying this paradigm to clear β-amyloid protein (Aβ) plaques and soluble aggregates from patients with Alzheimer’s disease (AD) is an extremely active field of research, with multiple active and passive Aβ vaccines currently in human clinical trials.[4] The most advanced of these is solanezumab/LY2062430 (Eli Lilly), which (along with gantenerumab and crenezumab) is one of three Aβ antibodies that are presently entering into late-stage trials in preclinical or early-stage AD. In the earlier posting, we surveyed research in applying this same paradigm to AS aggregates.
Since that post, there have been exciting developments in the progress of the two most advanced of these immunotherapies: PD01A, the AS-targeting active vaccine from Austrian biotechnology startup AFFiRiS AG, developed using its patented “AFFITOME” neo-antigen discovery platform of molecular mimicry; and PRX002, a humanized monoclonal Ab (mAb) under development from Prothena Corp PLC, the successor of aggregate-clearing immunotherapy pioneer Élan Pharmaceuticals. In the ensuing months, the two companies have published detailed, promising animal studies of their immunotherapies, including preclinical efficacy studies in animal models of human synucleinopathies. And both immunotherapies are now advancing through early-stage human clinical trials.
PD01A: Promising Animal Efficacy; Initial Human Safety and Hints of Disease Modification
At the time of our last post on α-synuclein vaccines, AFFiRiS had published several papers about the AFFITOME immnotherapy discovery platform[8-11] and had even begun their first Phase I trial of PD01A, but had kept nearly silent on many of the most elementary aspects of the vaccine. Such features as its actual target antigen were revealed only inconsistently and through secondary sources (such as a series of Michael J Fox Foundation blog posts and a third-party review article[14]), and only hints of their preclinical studies with the agent were disclosed (mostly [9,11]). In the summer of 2014, with their Phase I trial either complete or very nearly so, they released the first report on PD01A’s design, characteristics, and very encouraging animal model proof-of-concept results.[15]
Initial Design and Screening
AN1792, the first Aβ-targeting immunotherapy to be tested in humans, initially seemed highly promising. It exhibited safety in animal studies, and preliminary evidence of both target engagement and clinical efficacy in its Phase I and Phase II trials. But it was ultimately abandoned after a reformulated version used in the Phase II trial triggered meningoencephalitis in some 6% of patients.[13] AN1792 was an active vaccine, with Aβ aggregates as the antigen, and the meningoencephalitis is postulated to have been the result of the vaccine having roused a T-cell response against Aβ along with the necessary B-cell response, leading to a Th1-biased inflammatory attack centralized on sites in the brain with local Aβ deposits.[13] Highlighting this risk in the PD/AS context, a preclinical study using a nitrated AS species similar to one found in patient LB as an active vaccine antigen developed worrisome autoimmunity.[12] In fact, the investigators provided significant evidence that such an autoimmune reaction also occurs to a lesser extent in unvaccinated animals in the MPTP model of PD, and is itself responsible for part of the characteristic DA neuron loss in that system.[12]
As explained in our previous post, the AFFITOME system used to design PD01A seeks to avoid the risk of autoimmunity by exploiting the principle of molecular mimicry to identify novel, non-physiological peptides for use as antigens in active vaccines against physiological proteins and their aggregates. Molecular mimicry is the tendency of the immune system to cross-react from a primary target antigen to other, unrelated peptides (“neo-epitopes”) with distinct amino acid sequences and/or similar conformational structures.[8-11] In the AFFITOME system, antibodies specific to the physiological target are used to screen peptides whose amino acid sequences are unrelated to the original target antigen (and also, ideally, to other physiological antigens). In addition, the candidate pool is restricted to peptides with a sequence too short (≤8 amino acids) to bind to MHCI/II molecules, as is necessary for the induction of cell-mediated specific immunity. These two negative selections reduce the risk of an specific T-cell reaction to a physiological CNS protein that could lead to devastating autoimmune responses.[8,11,15]
To identify a candidate AS AFFITOME vaccine antigen for initial animal testing, the investigators used mABs targeting the C-terminus of AS to screen phage display libraries, seeking peptides to which such Abs would bind (and thus potentially capable of inducing the generation of Abs targeting the original AS terminus when used as vaccine antigens). The group excluded peptides from their screening pool if they harbored sequences homologous to those in AS itself, or to sequences present in other members of the AS family (β- and γ-synuclein).[15] β-synuclein was a protein of particular concern: it exhibits strong sequence homology with AS and a similar expression pattern, and yet it is not found in LB, is not amyloidogenic, and indeed appears inhibit AS aggregation and exhibit other neuroprotective actions.[5,6] Thus, cross-reactive autoimmunity against β-synuclein might be particularly deleterious, inducing an autoimmune attack on brain neurons while depleting them of a key constituent of their neuroprotective machinery.
From this initial screen, 7 candidate antigens emerged. These were then formulated into experimental vaccines by conjugation to keyhole limpet hemocyanin as a carrier peptide and aluminum hydroxide as an adjuvant. The original C-terminal AS peptide, with or without the formulants, were also used as control vaccine agents.[15] The inital pool of 7 candidates were then narrowed down through vaccination of both wild-type (WT) mice and of one of two transgenic models of human AS synucleinopathy (infra). To pass this secondary screen, candidates had to fulfil several criteria. They would have to elicit Abs in vaccinated animals that would bind to human AS, but not to murine AS or to human or mouse β-synuclein. Those inducedAbs would epitope map to recognize the C-terminus of AS as well as full-length and N-terminal truncated AS species, and also bind to oligomeric AS aggregates. And those same Abs would have to detect intracellular and axonal SN aggregates present in brain slices from synucleinopathic mice, but not to neurons from WT mice in vivo.[15]
From this series of screens, two candidates seemed to show initial promise, and were put through a series of additional screens to uncover any potential for potentially ruinous autoimmune T-cell reactions. Compared with either the carrier system alone or with unmodified human AS, neither candidate antigen primed T-lymphocytes derived from WT mouse spleen. Additionally, splenocytes derived from transgenic animals that had been immunized with one or the other candidate did not activate an AS-specific T-cell reaction following in vitro stimulation with AS or the candidate antigens themselves. And CD4+ T-cells in the blood vessels of vaccinated mice were rare, contrasting with experimental autoimmune encephalomyelitis (EAE) mice, which are a positive control model of T-cell-mediated CNS autoimmunity.[15]
Of the seven tested antigens, one emerged as a strong candidate vaccine: AFF 1. This was then subjected to initial testing for immunological efficacy in mThy1-AS mice, which overexpress WT human AS under the murine Thy1 promoter, and which exhibit sensorimotor behavioral deficits, axonal AS pathology, and C-terminally-truncated AS aggregates with regional distribution similar to PD and PD dementia (PDD).[16] Vaccination of these animals with AFF 1 elicited high titers of AS-targeting Abs, with substantial trafficking of Abs into cerebrospinal fluid (CSF), reaching 0.1-0.3% of plasma levels. Also importantly, no β-synuclein-targeting Abs were elicited. Furthermore, labeled mABs derived from treated mice and subsequently injected into mThy1s mice bound to human-derived AS aggregates in the neuropil and neuronal cell bodies in vivo, whereas no binding was observed in WT animals.[15] Thus, AFF 1-induced Abs not only reach the CSF but penetrate the blood-brain barrier (BBB) and are taken up selectively by neurons bearing human AS aggregates.
AFF 1: Preclinical Efficacy Data
AFF 1 was then tested for preclinical efficacy in two models of human synucleinopathy: mThy1-AS mice, and also PDGF-AS mice expressing WT human AS under the PDGF-β promoter — a model with closer neuropathological and metabolic resemblance to LBD than PD.[18]. Treatment with AFF 1 or vehicle control was initiated in mThy1-AS mice at age 3 mo, when early sensorimotor deficits first appear — similar to the target population for the first rollout of the human vaccine. By the age of 9-10 mo, neurpathology was advanced in vehicle-treated animals, but it was clear that AFF 1 had afforded substantial protection to treated mice. As compared to vehicle-treated mice, AFF 1 vaccination reduced the accumulated burden of SN AS pathology by 75%, and striatal neuropil AS aggregates by ≈45%. Examination of brain homogenates from immunized TG mice revealed no effect on monomeric human AS as compared with vehicle-treated animals, but a 45% reduction in oligomeric AS and a trend toward reduction of human AS dimers. There was no significant reduction of total human AS in brain homogenates, perhaps because the aggregates constitute so small a percentage of the total burden. Accompanying this, vaccination with AFF 1 prevented 66% of the loss of tyrosine hydroxylase-staining (TH+ — likely dopaminergic) neurons suffered by vehicle-treated mThy1-AS mice (all Figure 1).[15]
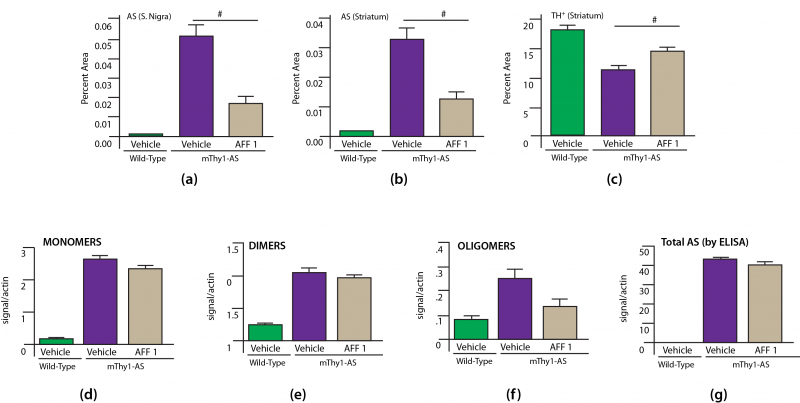
Figure 1. Effects of AFF 1 Vaccination on Neuropathology in PD-Moldel mThy1-AS mice. # = p < 0.05. Redrawn from [15] by Anne Corwin.
Motor dysfunction was evaluated in mThy1-AS mice using a modification of the established “coat hanger test:”[17] animals were assessed for time to fall when holding on to a suspended vertical metal bar, with additional scoring for the ability to use the hindlimbs to assist (zero, one, or both hindlimbs engaged).[15] While WT controls were able to hold onto the bar for 53 s in this test, vehicle-treated mThy1-AS mice lost hold of the bar in a mere 3.1 s on average.[15] AFF 1 vaccination preserved sufficient functionality to allow treated model PD mice to cling on for 19 s. Hindlimb engagement is by analogy a, particularly suggestive outcome for animal models of a disease of fine motor control impairment. mThy1-AS controls were unable to recruit even one hindlimb to prevent their own falling, but AFF 1 treatment maintained such engagement to half of WT control animals’ levels (average ≈1.0 vs. ≈1.75 hindlimbs engaged — all Figure 2).[15]
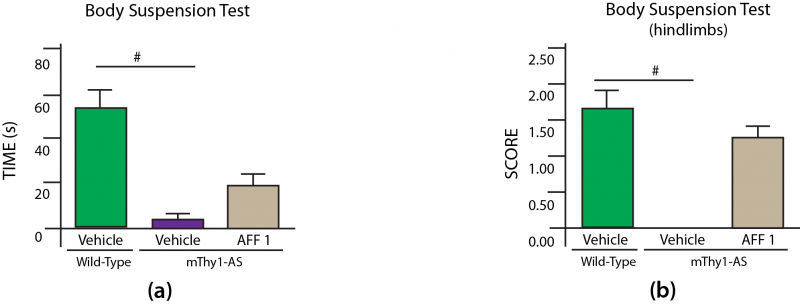
Figure 2. AFF 1 Ameliorates Neuromuscular Deficits in Model PD Mice. # = p < 0.05. Redrawn from [15] by Anne Corwin.
The investigators also tested the effects of AFF 1 in mice in the PDGF-AS mouse model of LBD. In this model, too, vaccination with AFF 1 induced high Ab titers in plasma, a meaningful percentage of which migrated into the CSF. Again, labeled mABs derived from AFF 1-vaccinated mice reached the brain parenchyma following systemic administration and colocalized with intraneuronal human AS. Consistent with this, AFF 1 immunotherapy reduced the accumulation of AS neuropathology in neuronal bodies and neuropil at the cardinal sites for this LBD model (neocortex and hippocampus), and reduced the levels of AS dimers as well as oligomers in brain homogenates, while having no significant effect on monomeric AS.[15] Significant recalcitrant (proteinase K-resistant) AS aggregate accumulated in the neuronal synapses of untreated PDGF-AS mice, as indicated by colocalization with synaptophysin staining; AFF 1 treatment significantly cleared this depot as well (Figure 3(d)).
In the process of reducing AS aggregate accumulation, vaccination with AFF 1 also afforded substantial protection against neurodegeneration per se. PDGF-AS mice suffered substantial dendritic atrophy and synaptic loss, as indicated by reductions in MAP2 and synaptophysin, along with a more modest loss of NeuN-staining neurons; treatment with AFF 1 almost completely normalized all three marker proteins, indicating preservation of neurons, dendrites, and synapses (Figure 3).
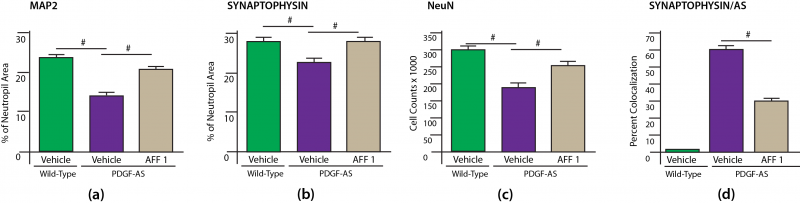
Figure 3. AFF 1 Preserves Synaptic Integrity. Redrawn from [15] by Anne Corwin.
Neuroinflammation, with activation of microglia and astroglia, is a prominent feature of the aging and PD brain. Microglial activation is an early event in PD that spreads to all disease-affected regions of the brain.[19] Culture studies show that recombinant AS activates microglia, leading to the death of cocultured DA neurons.[19] One of the effectors of microglial-mediated neuronal death is inducible nitric oxide synthase (iNOS), which is induced in PD brain and inflammatory-biased (M1) macrophages (including activated microglia) and reactive astrocytes.[19] In the current study,[15] PDGF-AS mice exhibited an increased number and intensity of areas staining for reactive microglia (using Iba1) and astroglia (using GFAP), but vaccination with AFF 1 prevented nearly all of this disease-associated excess, with treated animals exhibiting only the burden of reactive glia present in similar-aged WT mice. Similarly, iNOS expression levels were elevated in vehicle-treated PDGF-AS mice, but held to WT levels in animals receiving AFF 1 vaccine. Accompanying these abrogated neuroinflammatory changes in the brain, the levels of multiple anti-inflammatory cytokines were elevated in treated- vs. untreated mice.[15]
In addition to the reduction in total numbers of activated microglia in AFF 1-treated TG mice, it is of note that costaining studies indicated that those activated microglia that did emerge in treated mice were highly colocalized with recalcitrant (proteinase K-resistant) AS aggregate as compared to untreated PDGF-AS mice. (WT animals were free of recalcitrant aggregate).[15] The investigators hypothesize that the selective concentration of activated microglia with recalcitrant aggregates indicates that vaccination with AFF 1 facilitates microglial targeting and clearance of AS — a mechanism consistent with findings by Prothena-affiliated scientists using AS-targeting mAbs.[20,21] If validated, this model would be of particular interest because of the very strong evidence that the sequential sequential spread of AS pathology from the peripheral nervous system into and across the brain occurs via an “infectious” transynaptic spread from one neuron to the next.[28-30] Hence, microglial capture and degradation of extraneuronal AS aggregates might not only prevent the direct harms of extracellular AS aggregates, but potentially interrupt the cell-to-cell propagation of intraneuronal pathology and the progression of the disease.
For a functional test of the effects of AFF 1 vaccination in this model of AS-associated dementia, the investigators used the classic water maze trial. The effect of AFF 1 in PDGF-AS mice were even more marked than they had been in the more PD-like mThy1-AS mice: treatment completely abrogated the effects of AS transgenes on both time to reach the platform on the escape latency test (Figure 4a) and time in the target region when the platform was withdrawn in the probe test (Figure 4b).[15]
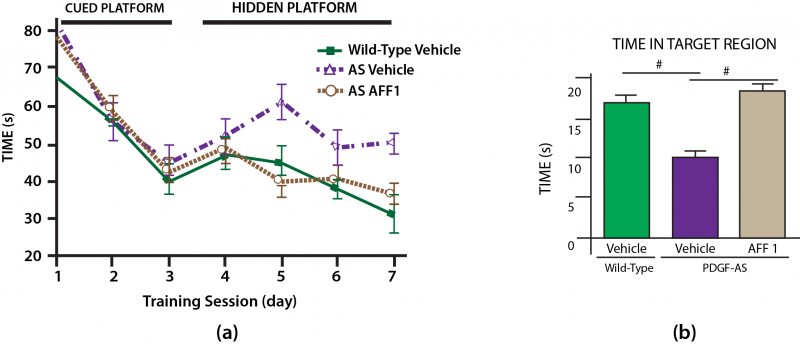
Figure 4. AFF 1 Prevents Cognitive Deficits in a Mouse Model of Lewy Body Dementia. Redrawn from (15) by Anne Corwin.
All in all, these were very encouraging preclinical results, and AFF 1 was advanced to human testing as PD01A.
PD01A in Phase I: Early Evidence of Safety and Hints of Efficacy
The animal data reviewed above were published in June of 2014,[15] long after they had evidently been completed (see e.g. [11]). As reviewed in our previous post, AFFiRiS announced the initiation of the first Phase I clinical trial of PD01A in June of 2012. This was a randomized, single-blind, multiple-dose, parallel-group controlled trial in 32 relatively young persons suffering with the early stages of PD (Hoehn and Yahr stage I-II). Patients were required to have been stable on PD medication for at least 3 months before study entry.[5]
Two groups of 12 subjects each were randomized to receive either a low (15 µg) or a high (75 µg) dose of the vaccine, one injection monthly for the first 4 months, and were monitored for a total of one year; 8 subjects serving as untreated controls and treated with best usual care. Patients were assessed in visits intermediate between each injection, in treated subjects on multiple safety, immunological, and potential clinical effects.[5] The primary outcome of the study was safety and tolerability, with immunological response to the vaccine as a secondary outcome. Subjects were also assessed on clinical endpoints using the Movement Disorder Society revision of the Unified Parkinson’s Disease Rating Scale (MDS-UPDRS), and underwent brain MRI, dopamine transporter (DaT) SPECT imaging, and lumbar puncture for evidence of anti-AS antibodies in CSF.
On July 31 of this year, AFFiRiS announced the auspicious results of this first trial. The different arms of the trial were very well-matched as regards age (average age ≈55 y, ± 5-7 y depending on subgroup), education, time from onset of disease, and MDS-UPDRS scores at onset; the only evident differences were a higher number of males and higher body weight in the low-dose treatment group. There were no dropouts in any group, and all active patients received all four scheduled injections and completed the trial.[5]
At the end of the one-year trial, all signs pointed to good short-term safety and tolerability. Particular effort was made to monitor subjects for signs of autoimmunity, and reassuringly none was observed; in particular, there was no sign of either treatment-associated autoimmunity or meningoencephalitis.[5] The adverse events that were observed over the course of the trial were equally distributed amongst the two dosed groups, with the single most frequent by far being typical local and self-limited injection-site inflammation.[5] Of the remainder, only three were serious adverse events, none of which were judged by the investigators to be unexpected or related to therapy: all occurred in the low- rather than the high-dose vaccine group, and all were lower back or knee pains common in older adults. The independent safety monitoring board agreed: by an unanimous vote, they judged there to be no serious safety or tolerability concerns raised by the trial.[5]
In terms of biological response: of 24 immunized subjects, 15 exhibited detectable antibody titers against AS, with titers ramping up progressively with each additional session of vaccination and then declining slowly over the remaining 8 mo, exhibiting a tendency toward a plateau at 40 weeks with titers at levels intermediate to those originally reported at the second and third immunizations ([5], and Figure 5). Importantly, AS-targeting Abs were also detectable in the CSF of 6 of 17 analyzed subjects,suggesting target engagement.[5]
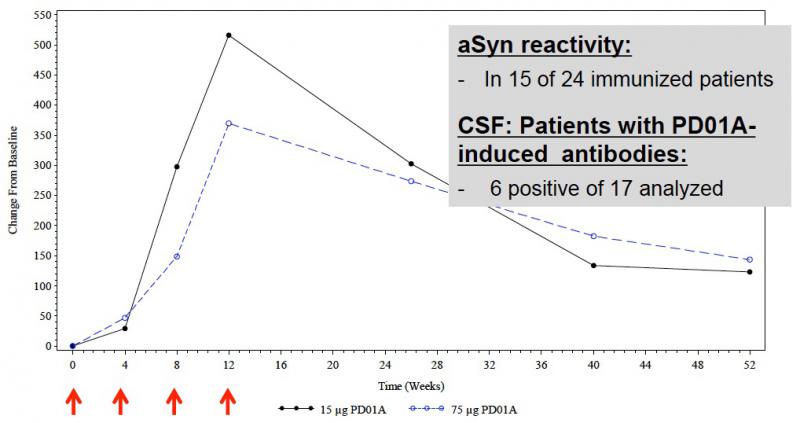
Figure 5. Immunologic Response to PD01A in Phase I Clinical Trial. Per-protocol analys of Abs targeting AS species. Reproduced from [5].
While not designed or powered as an efficacy study, there were also encouraging hints of clinical benefit in PD01A-treated patients. During the first 6 months after randomization, all participants’ investigator-rated global assessment (IGA) remained largely stable. But for every month thereafter, the control patients’ presentations precipitously declined, while both groups of volunteers receiving PD01A remained stable (Figure 6). The divergence from control subjects was statistically significant (15 µg dose) or nearly so (75 µg dose).

Figure 6. Investigator Global Assessment in PD01A-Treated Subjects and Controls. Reproduced from [5].
The results were even more auspicious when the investigators stratified vaccinated subjects by antibody responder status: subjects who successfully mounted an immunological response to the vaccine enjoyed statistically significant benefits as measured with the IGA, the two motor function subscales of the MDS-UPDRS, and the Parkinson’s Disease Questionnaire (PDQ-39) of quality of life (though not on the PD Non-Motor Symptoms Questionnaire (NMS-Quest/PD-NMS)), while no benefit was observed in nonresponders on any scale (Table 1).[5] It is not clear whether a “benefit” here constitutes an actual improvement from baseline status on these assays (so that the stable IGA illustrated in Figure 6, for instance, might be a composite of a positive amelioration of condition for responders while nonresponders continued to decline), or simply maintenance of stable disease status vs. a decline in nonresponders, but the result is in either case highly encouraging.
| Nonresponders | Responders | p-value |
n | 17 | 15 |
|
IGA | – | Benefit | ≤0.05 |
UPDRS-II | – | Benefit | ≤0.05 |
UPDRS-III | – | Benefit | <0.001 |
PDQ-39 | – | Benefit | ≤0.05 |
PD-NMS | – | – | n.s. |
It is puzzling that immunological responders seemed to benefit in all measures except the nonmotor symptoms: mechanistically, it is these that we would most expect to be ameliorated or improved, particularly in the early stages of the disease. However, as AFFiRiS Chief Medical Officer Achim Schneeberger rightly emphasized when asked at the press conference,[5] the results seen to date are of a 1 y trial in just 24 subjects and with only 15 vaccine responders: it is greatly premature to conclude that PD01A will not deliver benefits in nonmotor domains.
Similarly, it is provocative, albeit speculative, to note in the lower dose of the vaccine seemed to yield a stronger response than the higher dose on several outcomes: Ab titer, IGA, and potentially even adverse reactions. On the other hand, this subgroup also included more males, and the participants were of higher weight at baseline, and it is conceivable that these population differences were causally linked to a the nominally stronger response. But this interpretation too is highly speculative in so small a group and on outcomes on which the study was not designed to provide reliable evidence.
Indeed, more broadly speaking, one must be careful not to place too much weight on any of these results, promising as they may seem prima facie, or to overinterpret them. In addition to the small sample size and the fact that the trial was not designed or intended as an efficacy study, one important caveat is that because the trial was not investigator-blinded, it is likely that at least some bias was introduced into the assessments, particularly on the more subjective ones such as the IGA and some components of the MDS-UPDRS II and III subscales. It is also unclear why results are not presented for the MDS-UPDRS Ia subscale or for cognition, which were both listed in the study protocol as secondary outcomes. It may be that there was an early decision to rely on the PD-NMS as the sole instrument for nonmotor symptoms of PD, replacing the MDS-UPDRS Ia and subsuming any separate cognitive instrument; it would be useful for the investigators to clarify this point, preferably in advance of formal publication of the trial.
Still, while these findings must be regarded as highly preliminary, we must agree with AFFiRiS that these results are, at minimum, consistent with a disease-modifying effect of PD01A — an exciting prospect, in a disease with effective symptomatic treatments but no disease-modifying therapies, and a course as inexorable as the degenerative aging processes that drive it.
PD01A — and PD03: Passing the Baton, and a New Race
As announced at the press conference, AFFiRiS is now inviting all subjects from the original Phase I trial to participate in a followup Phase Ib booster study. All subjects that volunteer for the booster study will have their clinical status reassessed; two months later, volunteers that had previously received vaccine during the original trial will be given a booster injection of PD01A. The booster dose will be randomized, such that some subjects previously receiving 15 µg will receive 75 µg as their booster and vice-versa. Control subjects will continue to receive best practice medical care, but no injection. Safety, immunologic, and clinical outcomes will be similar to those in the original trial; subjects will be followed for a total of 6 additional months.
Curiously, the Clinicaltrials.gov website also lists a 1 y, purely observational extension study of the original trial, with overlapping timelines and patient numbers to the booster study, during which no subject will receive further treatment. The stated enrollment goals suggest the intent to recruit all subjects from the original trial into both studies, which appears to be impossible per the stated protocols. One might speculate that this will be for subjects who refuse the booster study, and that the stated ns for the two trials are maxima rather than actual targets.
Meanwhile, the European Union-funded SYMPATH Project has received nearly €6 million from the Seventh Framework Program to test AFFiRiS’ AS-targeting vaccines in patients suffering with synucleinopathies. The clinical consortium is led by AFFiRiS itself, and in addition to the PD booster trial, it is now recruiting for a separate Phase I trial of the α-synuclein vaccine against MSA, a synucleinopathy differentiated from PD or LBD based on the cell populations affected, the regional concentration of AS neuropathology, and the spectrum of movement and other symptoms. MSA is also faster-progressing than PD, and is typically not responsive to levodopa, denying the afflicted even symptomatic relief from disturbing autonomic and motor nervous system dysfunction.
AFFiRiS aims to recruit 30 volunteers with MSA for the randomized, parallel-group, single-blind study, to be carried out in clinical centers in Toulouse and Bordeaux, France. Unlike the original PD01A PD trials, the MSA trial will use a biologically active but presumptively clinically inert (adjuvant only) placebo control. Over the course of a year, twelve subjects will receive a total of 5 injections of 75µg of PD01A: the first 4 injections administered once every 4 weeks, followed by a booster injection 36 weeks after the first. An additional 12 volunteers will instead receive 75µg of PD03A, a vaccine evidently related to PD01A but not otherwise characterized in public information identified by this author. Six additional subjects will serve as placebo (adjuvant only) controls. All subjects will be evaluated for safety, immunological, and preliminary clinical outcomes similar to those for PD but using clinical instruments suitable for MSA.
A Competitor Mounts the Starting Blocks
It would be exciting enough for one AS-targeting rejuvenation biotechnology (or one and a half, if PD03A is counted separately) were making such progress through the clinical pipeline, especially in light of the early hints of a disease-modifying effect. But now a rival vaccine is rapidly advancing on its heels. Having previously published data on a variety of experimental mAbs against AS species in animal models ([16,20]; possibly [21]), Prothena’s Dale Schenk presented new data on additional experimental antibodies at SENS Research Foundation’s Rejuvenation Biotechnology 2014 conference, roughly contemporaneous to its publication,[22] along with information specific to the humanized candidate PRX002, which is now being advanced into clinical trials.
Strategy: Passive Vaccination Against C-Terminally-Truncated α-Synuclein
Prothena have focused their development pathway on passive immunotherapy with mAbs targeting C-terminally-truncated AS species (CTTAS), which accumulate in neurons and neuropil in PD and LBD brain,[23] selectively in the neuropil of AD brain with or without neuronal LB,[24] and (usefully) in the mThy1-AS mouse.[16] C-terminal cleavage of AS promotes its oligomerization, cell-to-cell transmission, and toxicity (see citations in [16]). Expression of the familial PD-linked A53T or A30P mutant ASs in cell models results in an increased proportion of CTTAS being produced than is the case when expressing the WT protein; consistent with this, a higher proportion of the total soluble AS in human brain tissue with AS neuropathology is CTTAS, and a higher proportion yet of the total AS in insoluble AS deposits.[25]
In their most recent published report,[22] Prothena scientists prepared three new candidate mAbs, each targeting different sequences in the AS C-terminus. 1H7 was raised from recombinant full-length AS, as was 9E4, a mAb previously used successfully to target AS in PDGF-AS mice[20] and was therefore included in the new study as a positive control for the 3 novel mAbs. Additional mAbs (5C1, 5D12) were raised using AS peptides. They also included non-specific IgG as a control treatment. Of these, 1H7, 5C1, and 9E4 would prove to be effective in numerous in vitro and in vivo tests. By contrast, while 5D12 was not designed as a negative control, it proved significantly less effective than the other candidates, for reasons that are not entirely clear.
Amongst other mechanistically-significant findings, 1H7, 5C1, and 9E4 recognized immunoblot bands in brain homogenates from WT and mThy1-AS mice that were consistent with AS monomers, potential oligomers, and a CTTAS species, while 5D12 recognized monomers and a smaller set of potential oligomers. And while all four antibodies immunostained mThy1-AS mouse brain neuronal soma and synapses, with 1H7 being the most potent, further studies showed that while 1H7, 5C1, and 9E4 stained for AS species in both soma and neuropil, 5D12 staining with AS was largely restricted to the soma.[22]
The lack of 5D12 binding in synaptic zones may limit its therapeutic effect, as appeared to be the case in LBD-model mice (vide infra). In PD and particularly LBD brain, AS aggregates are found at autopsy in both the soma (LB) and neuropil (Lewy neurites), with a higher concentration of Lewy neurites in LBD. Moreover, cell-to-cell transmission of “infectious” AS via the synapse is emerging as an important candidate mechanism for progression of AS pathology,[28-30] making antibody-mediated clearance of secreted/exocytosed AS a potential mechanism of interrupting the neuropathological spread. Such therapeutic interdiction was achieved by these investigators in previous work in vivo with 9E4[20] and other C-terminus-directed AS-targeting Abs,[21] and in the current study the investigators found that 1H7, 9E4, or 5C1 largely interrupted the cell-to-cell propagation of AS out of neuroblastoma cells transgenically overexpressing intact AS, while 5D12 exerted minimal effects.[22]
Additionally, 1H7, 9E4, and 5C1 were all more effective than 5D12 at in vitro inhibition of C-terminal cleavage of recombinant AS by calpain-1.[22] This may be a further mechanism of disease modification, as calpain-1 has emerged as the most likely of several enzymes for the generation of pathologically-relevant CTTAS from AS in vivo.[16,23,27]
Prevention of CTTAS Neuropathology, Neurodegeneration, and Behavioral Deficits
Like AFFiRiS scientists testing AFF 1,[15] the Prothena group selected the mThy1-AS mouse for in vivo testing of their candidate mAbs. As previously noted, these animals accumulate CTTAS in vulnerable brain regions in a pattern consistent with that observed in humans suffering with diagnosed PD and LBD,[16] making them a good model for testing mAbs intended to target these species. (As also noted previously, 9E4 — used as a positive control in this study[22] — had already been shown effective in PDGF-AS mice[20]). Animals were administered 10 mg/kg of candidate mAbs weekly for 6 mo; control animals (WT and TG) were immunized with IgG1 as a control Ab.
Compared to control mThy1-AS mice, treatment with 1H7, 5C1, and 9E4 reduced total AS species in the neuropil of the temporal cortex, and CTTAS aggregates in striatal and temporal cortex neuropil; there was also a trend toward reducing total intraneuronal AS in the temporal cortex of animals treated with 1H7 or 5C1. 5D12 was ineffective in all of these outcomes, although it did reduce CTTAS in the neuropil of the neocortex (Figure 7). Consistent with these results, colabeling experiments with mAbs against neurofilament as a marker for axons and against CTTAS showed that 1H7, 9E4, and to a lesser extent 5C1 reduced the percentage of striatal axons occupied by CTTAS, while 5D12 was again without significant effect. Importantly, none of these Abs bound to native mouse AS in WT animals, consistent with their intended selectivity for human-derived AS species.[22]
Prothena’s candidate vaccines seem to have had no effect on the relatively mild loss of SN DA neurons in this model, but to have afforded substantial protection against the more profound losses in the striatum. Treatment of mThy1-AS mice with 1H7 or 9E4 (and to a lesser extent 5C1) reduced the percentage of striatal axons burdened with CTTAS; 5D12 was ineffective in this regard. mThy1-AS mice administered only control IgG1 suffered dendritic atrophy (as suggested by reduced staining with MAP2) and synaptic loss (evaluated with postsynaptic density-95 (PSD-95) and synaptophysin) across several regions of the brain, but this neurodegenerative damage was substantially prevented in mice administered 1H7, 5C1, or 9E4. The effects of 5D12 varied depending on brain region. (For graphical presentation of all but MAP2 data, see Figure 7).

Figure 7. PRX002 Protects Against Neurodegeneration Driven by α-Synuclein Aggregates in a Mouse Model of Parkinson’s Disease. Redrawn from [22] by Anne Corwin. Synaptophysin in (f) by immunoblot by in brain homogenates; for regional effects on synaptophysin and MAP2, analyzed in serial sections by immunohistochemistry, see Fig. 8 in [22]).
As with AFFiRiS’ active vaccine, passive vaccination with 5C1, 9E4, or 1H7 afforded substantial protection against the emergence of neuroinflammation, evidenced by ameliorated GFAP staining for reactive astrocytes and Iba1 for microgliosis. 1H7 provided particularly strong protection against astrocytosis, while 5D12 provided no protection against the former and only a nonsignificant trend toward protection against the latter (Figure 7).
The investigators then tested the effects of their candidate mAbs on functional assays in their mThy1-AS mice. As expected, transversal round beam testing[26] revealed substantial deficits in motor function in this PD model. All four candidates appeared to substantially prevent the emergence of these deficits, although only the effects of 1H7 and 9E4 crossed the threshold of significance when compared with other mThy1-AS mice administered control IgG. Surprisingly, in fact, the performance of animals treated with either 1H7 or 9E4 was nominally superior than that of similarly-aged WT animals, although the differences were again not significant. At minimum, these two antibodies were clearly sufficiently powerful as to completely obviate the behavioral deficits exhibited by untreated PD-model mice (Figure 8).[22]
Cognitive function was also assessed, using the water maze.[22] AFFiRiS had used water maze testing to evaluate AFF 1’s ability to ameliorate cognitive deficits in PDGF-AS mice,[15] and Prothena investigators had similarly demonstrated the efficacy of 9E4 in the water maze using this model.[20] Here they extended this finding to mThy1-AS mice. The PD model animals did prove to exhibit substantial water maze deficits, both in time to locate the hidden platform and in time spent in the target quadrant on the probe test. Remarkably, treatment with 1H7, 9E4, or 5C1 were each able to completely or near-completely prevent the emergence of these deficits, with treated animals performing at levels not significantly different from those of their WT controls. By contrast, 5D12 was largely ineffective (Figure 8), consistent with its weaker or nonexistent effects against several neuropathological and neurodegenerative outcomes.[22]
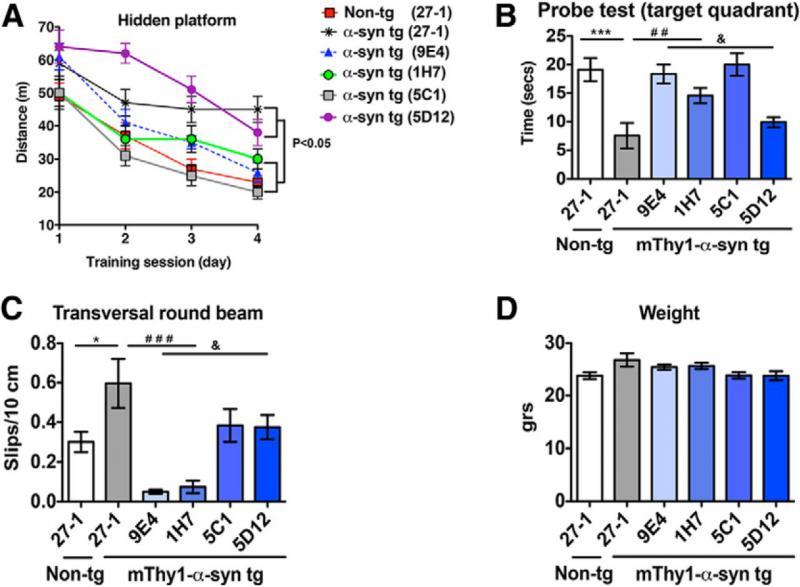
Figure 8. Prothena Passive Vaccines Ameliorate Cognitive and Motor Deficits in Model Parkinson’s Mice. ©2014 Society for Neuroscience. Reproduced from [22], with permission.
Advance to the Clinic
On December 11, 2013, presumably on the strength of these sweeping (but then-unpublished) preclinical data, the pharmaceutical giant Roche announced:
Roche (SIX: RO, ROG; OTCQX: RHHBY) and Prothena Corporation plc (NASDAQ:PRTA), announced today that they have entered into a worldwide collaboration to develop and commercialize antibodies that target alpha-synuclein, including PRX002 … which is currently in preclinical development and is expected to enter Phase 1 clinical trials in patients with Parkinson’s disease in 2014. …
Roche and Prothena will collaborate on the development of PRX002 for Parkinson’s disease and potentially other synucleinopathies. … Prothena will receive an upfront payment and near-term clinical milestone totaling USD45 million. Prothena is also eligible to receive additional payments of up to USD380 million upon the achievement of development, regulatory and first commercial sales milestones plus up to an additional USD175 million in ex-U.S. commercial milestone payments. The total worldwide upfront and milestone payments may amount up to USD600 million.
Also as part of the agreement, Roche and Prothena will initiate a research collaboration focused on optimizing early stage antibodies targeting alpha-synuclein including incorporation of Roche’s proprietary Brain Shuttle™ technology to increase delivery of therapeutic antibodies to the brain [via the transferrin receptor].
The partners worked quickly to move PRX002 into clinical trials. According to Prothena, “An Investigational New Drug application, or IND, for PRX002 was filed and accepted by the FDA in March 2014. Together with Roche, we initiated a Phase 1 clinical trial for PRX002 for the treatment of Parkinson’s disease in April 2014.” Within monts, PRX002 became the subject of two randomized, double-blind, placebo-controlled clinical trials: a three-month single-dose trial (Phase Ia) and a six-month multiple-dose ascending trial (Phase Ib). The shorter-term single-dose trial is recruiting 40 healthy and relatively young (ages 21-65 y) subjects for a narrow safety trial, whereas the multiple-dose trial will be conducted in 60 persons suffering with PD (the trial protocol specifies the recruitment of idiopathic PD subjects), and includes subjects that are somewhat older (ages 40-80) and with a wider range of severity of disease (Hoehn and Yahr stages I-III) than were used in AFFiRiS’ trial of PD01A. The primary outcomes of both trials are safety (adverse events) tolerability, and a wide array of pharmacokinetic outcomes (which are curiously designated as safety issues). One secondary outcome will be immunogenicity, meaning monitoring subjects for any emergence of antibodies against the infused therapeutic antibodies themselves; additionally, “Multiple clinical and exploratory biomarkers will be assessed” in the Phase Ib trial.
Prothena announced the dosing of the first Phase Ia subject on April 8, 2014; they appear to have been successful in recruiting subjects and to have quickly derived favorable safety signals, as they announced the dosing of the first PD volunteer in the Phase Ib trial on July 31, 2014, “based upon safety and tolerability observed to date in the ongoing study in healthy volunteers.”
As we have seen, both active vaccination with AFFiRiS’ AFFITOPE AFF 1/PD01A vaccine and passive vaccination with Prothena’s AS-targeting mAbs (one of which presumably was humanized to develop PRX002) were effective in rescuing neuronal loss, synaptic degeneration, neuroinflammation, and cognitive and behavioral deficits in two mouse models of synucleinopathies of aging. There is no direct way to compare the separately-reported effects of the two approaches even in mice, although it may be significant that AFFiRiS’ results were achieved after AFF 1 treatment was initiated in mice starting at 3-4 mo of age, when only the earliest signs of PD-like behavioral alterations have appeared,[15] whereas Prothena’s test animals were somewhat older (6 mo) — and thus presumably suffering with a greater burden of neuropathology and disability at the onset of treatment.[22]
Prospects in Competition and Cooperation
The entry of these two AS-targeting rejuvenation biotechnologies into human clinical testing would seem to mark an inflection point. As recently as two years ago, there was little evidence of any academic or biotech industry interest in pursuing clearance of AS as a therapeutic approach to PD, LBD, or other synucleinopathies of aging: after a promising 2005 report,[31] there had been virtual silence. Today, two AS aggregate-clearing immunotherapies have advanced into human testing, with one using an active vaccine approach and the other a passive mAb infusion strategy. Either or both may prove effective. One might speculate that each of these damage-repair therapeutics may exhibit differential targeting of particular AS species or brain regions, as different clinical synucleinopathies exhibit distinct regional localizations and conformations of AS pathology.
We note here an example of the perverse siloing of the various subfields of rejuvenation biotechnology that emerged during the question period that followed the surprisingly reserved delivery of the exciting news at the AFFiRiS press conference.[5] Having just presented the results of their first-in-human clinical experience with an AS aggregate-targeting vaccine, an emailed question asked if the investigators expected that PD01A would still be effective in patients who had progressed beyond the early stages of PD. Schneeberger said that what he and many others “have seen” is that in PD, as in AD, “once the the brain cells are destroyed, getting them back is probably impossible. So it’s really something we need to apply early in this disease.” He gave a similarly negative reply to a question about the use of the vaccine in patients undergoing deep brain stimulation (DBS) for PD, “given the mode of action of disease modifiers,” because DBS is typically reserved to patients in relatively advanced stages of the disease.
Schneeberger’s statements are consistent with views often expressed by researchers pursuing immunotherapy-based damage-repair prophylaxis of neurodegenerative aging disorders. And they have a sound grounding in the data — when considering aggregate clearance as a monotherapy, in isolation from developments in other areas of rejuvenation biotechnology. Both recent experience with immunotherapy for clearance of Aβ in AD, and their own (and Prothena’s) experience with AS-clearing immunotherapies in animal models, indicate that in order to be effective as disease-modifying agents when administered alone, therapies that remove proteinaceous aggregates from the brain must be initiated in the early clinical or even preclinical stages of the disease, before the burden of other forms of aging damage becomes entrenched.
These findings are entirely in line witht the known etiopathogenesis of the neurodegenerative diseases of aging. AD, PD, and other diseases and functional deficits of degenerative aging processes emerge from the ongoing and often mutually-reinforcing accumulation of multiple forms of cellular and molecular damage. In the case of PD, the most prominent and causally proximate forms of aging damage driving the disease are AS neuropathology and loss of DA neurons from the SN, but there is more. Large deletions in mitochondrial DNA are especially prominent in aging and PD SN DA neurons,[32,33] and contribute to the dysfunction and perhaps the loss of such neurons.[32-34] And recently, emerging evidence has pointed to an important indirect role played by senescent cells (specifically, senescent glial cells) in PD, contributing to the dysfunctional neuroinflammation and SN DA neuron loss in PD,[35] and researchers are now reporting the salutary effects of their therapeutic clearance.[36]
But while substantial data (including AFFiRiS'[15] and Prothena’s[20,22] preclinical work) give good reason to think that early clearance of AS neuropathology will decelerate the rate of SN DA neuronal loss, astrocytosis, and possibly accumulation of senescent glial cells in PD, there is no reason to think that doing so will prevent these phenomena entirely, if for no other reason than because many other factors also contribute to their occurrence. Additionally, there is only very limited and somewhat flawed evidence for neurogenic replacement of lost SN neurons,[37] so this is not an instance where one might anticipate that the repair of one form of damage in a tissue might open up space for endogenous repair systems to reverse categories of damage that are not the direct target of the original therapy.
But foreseeable rejuvenation biotechnology exists to repair and reverse this other aging damage. Allotopic expression of the mitochondrially-encoded proteins and/or allotopic mRNA can be anticipated to obviate the effects of mitochondrial mutations. The deleterious effects of senescent astrocytes can be expected to be reversed by therapeutic clearance (for which proof-of-concept already exists, including in the specific PD context[36]). And above all, mature cell therapy can be expected to restore the dopaminergic cellularity of the SN and ultimately innervation of the striatal targets, leading to recovery of DA signaling and the maintenance or regaining of motor control. Indeed, PD was one of the first diseases of aging in which a relatively crude form of cell therapy was used to replace missing cells and partially restore function in some patients.[38] Although the results of the trials of cell therapy for PD were mixed, with only a minority of patients showing clear-cut and durable gains, researchers have subsequently gained substantial insight into the reasons underlying differential outcomes at different centers and amongst different protocols, and for the troubling dyskinesias that were suffered by some patients.[38] And the most recent reports of long-term outcomes from several of the early cell therapy trials are much more promising than might have been expected from earlier reports, with transplanted cells surviving for as long as 14 y, exhibiting evidence of functional integration, and some patients continuing to further improve over the course of many years after engraftment.[40-42]
So when AFFiRiS’ Schneeberger opines that “given the mode of action of disease modifiers”, an AS-clearing immunotherapy such as PD01A (or PRX002) is “really something we need to apply early in this disease” because “once the the brain cells are destroyed, getting them back is probably impossible,” he is adopting a view that is entirely reasonable — but only because it is constrained to the specific plank in the integrated platform that rejuvenation biotechnology must become.
Fortunately, progress continues on developing new therapeutics aimed at removal, repair, replacement, or rendering harmless of all of the key lesions in the neurodegenerative aging processes that erode every aging person’s health and functionality and that progress into the clinical diagnosis of PD in an unfortunate few. In addition to AFFiRiS and Prothena, BioArctic Neuroscience AB continues to report earlier-stage but promising preclinical results with their lead candidate PD0805, a mAB targeting AS protofibrils.[43,44] As this blog post was being drafted, a collaboration between Masliah’s group and San Diego biotech startup NeuroTransit, located in Johnson & Johnson’s Janssen Labs incubator center, published successful preclinical results using yet another immunotherapy-based approach to AS clearance. By linking a recombinant single-chain fragment variable (scFV) targeting AS oligomers to the low-density lipoprotein receptor-binding domain of apolipoprotein B, they were able to facilitate uptake of the therapeutic hybrid protein across the BBB and thereafter the cellular uptake, lysosomal trafficking, and degradation of the ensuing scFV-AS complex. When administered intraperitoneally to PDGF-AS mice, this conjugate reduced AS neuropathology, neuronal loss, astrogliosis, and behavioral deficits in the water maze.[47]
Meanwhile, there is one human clinical trial already underway to build on the promise of the first faltering steps in cell therapy for PD, and several more trials are in planning. The TRANSEURO trial of fetal ventral mesencephalic cell grafts has already implanted cells in its first patient, using improved patient selection, cell sourcing, and graft techniques. The Summit4StemCell initiative aims to fund a trial of induced pluripotent stem cell (iPS)-derived DA neurons in persons in the early stages of PD through private charitable donations.[39] And two additional trials with iPS-derived neurons or progenitors are planned in the next 1-2 y: one by Jun Takahashi of Kyoto University in Japan (cf. here and here), and another being organized by Ivar Mendez (Chair of the Department of Surgery at the University of Saskatchewan) and Ole Isacson of Harvard.
Additionally, Mayo Clinic scientists and a multi-investigator collaboration centered around Judith Campisi and collaborators (including but not limited to Andersen[35,36]) are working on methods to ablate senescent cells from aging tissues.[45,46] And focusing on the critical-path work receiving the least investment from alternative government and industry sources, Matthew O’Connor’s team at SENS Research Foundation’s Research Center project on mitochondrial mutations continue their work toward obviation of these lesions, advancing both AE protein and mRNA strategies.
There is work to be done to advance all of these individual rejuvenation biotechnologies into and through clinical testing. Regulatory reform will be necessary to adopt a system based on treating existing pathology to one based on repair of the cellular and molecular damage that causes it, with the outcome being the delay and eventual indefinite postponement of the very onset of such pathology with age. And the greater work of catalyzing the transition of biomedical research generally from targeting metabolic pathways for “risk factor” reduction in favor of repair of the lesions that accumulate in a tissues, impair their function, and lead to morbidity and mortality must be realized.
The full potential of the “mode of action of disease modifiers such as PD01A” will not come through their isolated development, but through their integration into a protocol whose aim is not merely the piecemeal amelioration of individual clinical diagnoses, but the comprehensive enlivening of aging bodies and the rejuvenation of human lives.
References
1: Lim SY, Fox SH, Lang AE. Overview of the extranigral aspects of Parkinson disease. Arch Neurol. 2009 Feb;66(2):167-72. doi: 10.1001/archneurol.2008.561. Review. PubMed PMID: 19204152.
2: Stern MB, Lang A, Poewe W. Toward a redefinition of Parkinson’s disease. Mov Disord. 2012 Jan;27(1):54-60. doi: 10.1002/mds.24051. PubMed PMID: 22252891.
3: Pellicano C, Benincasa D, Pisani V, Buttarelli FR, Giovannelli M, Pontieri FE. Prodromal non-motor symptoms of Parkinson’s disease. Neuropsychiatr Dis Treat. 2007 Feb;3(1):145-52. PubMed PMID: 19300544; PubMed Central PMCID: PMC2654529.
4: Lobello K, Ryan JM, Liu E, Rippon G, Black R. Targeting Beta amyloid: a clinical review of immunotherapeutic approaches in Alzheimer’s disease. Int J Alzheimers Dis. 2012;2012:628070. doi: 10.1155/2012/628070. Epub 2012 Jan 15. PubMed PMID: 22292124; PubMed Central PMCID: PMC3265072.
5: Schmidt W, Sherer T, Schneeberger A. AFFiRiS AG Presents Early Clinical Data on a First-of-its-kind Parkinson’s Disease Vaccine. New York Marriott East Side Hotel, New York, NY. 2014-07-31. Accessed 2014-11-03.
6: Tsigelny IF, Bar-On P, Sharikov Y, Crews L, Hashimoto M, Miller MA, Keller SH, Platoshyn O, Yuan JX, Masliah E. Dynamics of alpha-synuclein aggregation and inhibition of pore-like oligomer development by beta-synuclein. FEBS J. 2007 Apr;274(7):1862-77. PubMed PMID: 17381514.
7: Hashimoto M, Rockenstein E, Mante M, Mallory M, Masliah E. beta-Synuclein inhibits alpha-synuclein aggregation: a possible role as an anti-parkinsonian factor. Neuron. 2001 Oct 25;32(2):213-23. PubMed PMID: 11683992.
8: Schneeberger A, Mandler M, Otawa O, Zauner W, Mattner F, Schmidt W. Development of AFFITOPE vaccines for Alzheimer’s disease (AD)–from concept to clinical testing. J Nutr Health Aging. 2009 Mar;13(3):264-7. Review. PubMed PMID: 19262965.
9: Schneeberger A, Mandler M, Mattner F, Schmidt W. Vaccination for Parkinson’s disease. Parkinsonism Relat Disord. 2012 Jan;18 Suppl 1:S11-3. doi: 10.1016/S1353-8020(11)70006-2. Review. PubMed PMID: 22166404.
10: Schneeberger A, Kutzelnigg A, Mandler M. Parkinson’s disease vaccine: clinical trial challenges when striving for disease modification. Clin Invest. 2013 Jan;3(1):1-4.
11: Schneeberger A, Mandler M, Mattner F, Schmidt W. AFFITOME® technology in neurodegenerative diseases: the doubling advantage. Hum Vaccin. 2010 Nov;6(11):948-52. Epub 2010 Nov 1. Review. PubMed PMID: 20980801.
12: Benner EJ, Banerjee R, Reynolds AD, Sherman S, Pisarev VM, Tsiperson V, Nemachek C, Ciborowski P, Przedborski S, Mosley RL, Gendelman HE. Nitrated alpha-synuclein immunity accelerates degeneration of nigral dopaminergic neurons. PLoS One. 2008 Jan 2;3(1):e1376. doi: 10.1371/journal.pone.0001376. PubMed PMID: 18167537; PubMed Central PMCID: PMC2147051.
13: Pride M, Seubert P, Grundman M, Hagen M, Eldridge J, Black RS. Progress in the active immunotherapeutic approach to Alzheimer’s disease: clinical investigations into AN1792-associated meningoencephalitis. Neurodegener Dis. 2008;5(3-4):194-6. doi: 10.1159/000113700. Epub 2008 Mar 6. Review. PubMed PMID: 18322388.
14: Hutter-Saunders JA, Mosley RL, Gendelman HE. Pathways towards an effective immunotherapy for Parkinson’s disease. Expert Rev Neurother. 2011 Dec;11(12):1703-15. doi: 10.1586/ern.11.163. Review. PubMed PMID: 22091596; PubMed Central PMCID: PMC3263336.
15: Mandler M, Valera E, Rockenstein E, Weninger H, Patrick C, Adame A, Santic R, Meindl S, Vigl B, Smrzka O, Schneeberger A, Mattner F, Masliah E. Next-generation active immunization approach for synucleinopathies: implications for Parkinson’s disease clinical trials. Acta Neuropathol. 2014 Jun;127(6):861-79. doi: 10.1007/s00401-014-1256-4. Epub 2014 Feb 14. PubMed PMID: 24525765; PubMed Central PMCID: PMC4034750.
16: Games D, Seubert P, Rockenstein E, Patrick C, Trejo M, Ubhi K, Ettle B, Ghassemiam M, Barbour R, Schenk D, Nuber S, Masliah E. Axonopathy in an α-synuclein transgenic model of Lewy body disease is associated with extensive accumulation of C-terminal-truncated α-synuclein. Am J Pathol. 2013 Mar;182(3):940-53. doi: 10.1016/j.ajpath.2012.11.018. Epub 2013 Jan 9. PubMed PMID: 23313024; PubMed Central PMCID: PMC3589076.
17: Brooks SP, Dunnett SB. Tests to assess motor phenotype in mice: a user’s guide. Nat Rev Neurosci. 2009 Jul;10(7):519-29. doi: 10.1038/nrn2652. Epub 2009 Jun 10. Review. PubMed PMID: 19513088.
18: Amschl D, Neddens J, Havas D, Flunkert S, Rabl R, Römer H, Rockenstein E, Masliah E, Windisch M, Hutter-Paier B. Time course and progression of wild type α-synuclein accumulation in a transgenic mouse model. BMC Neurosci. 2013 Jan 9;14:6. doi: 10.1186/1471-2202-14-6. PubMed PMID: 23302418; PubMed Central PMCID: PMC3546911.
19: Sanchez-Guajardo V, Barnum CJ, Tansey MG, Romero-Ramos M. Neuroimmunological processes in Parkinson’s disease and their relation to α-synuclein: microglia as the referee between neuronal processes and peripheral immunity. ASN Neuro. 2013 Apr 30;5(2):113-39. doi: 10.1042/AN20120066. Review. PubMed PMID: 23506036; PubMed Central PMCID: PMC3639751.
20: Masliah E, Rockenstein E, Mante M, Crews L, Spencer B, Adame A, Patrick C, Trejo M, Ubhi K, Rohn TT, Mueller-Steiner S, Seubert P, Barbour R, McConlogue L, Buttini M, Games D, Schenk D. Passive immunization reduces behavioral and neuropathological deficits in an alpha-synuclein transgenic model of Lewy body disease. PLoS One. 2011 Apr 29;6(4):e19338. doi: 10.1371/journal.pone.0019338. PubMed PMID: 21559417; PubMed Central PMCID: PMC3084838.
21: Bae EJ, Lee HJ, Rockenstein E, Ho DH, Park EB, Yang NY, Desplats P, Masliah E, Lee SJ. Antibody-aided clearance of extracellular α-synuclein prevents cell-to-cell aggregate transmission. J Neurosci. 2012 Sep 26;32(39):13454-69. PubMed PMID: 23015436; PubMed Central PMCID: PMC3752153.
22: Games D, Valera E, Spencer B, Rockenstein E, Mante M, Adame A, Patrick C, Ubhi K, Nuber S, Sacayon P, Zago W, Seubert P, Barbour R, Schenk D, Masliah E. Reducing C-terminal-truncated alpha-synuclein by immunotherapy attenuates neurodegeneration and propagation in Parkinson’s disease-like models. J Neurosci. 2014 Jul 9;34(28):9441-54. doi: 10.1523/JNEUROSCI.5314-13.2014. PubMed PMID: 25009275; PubMed Central PMCID: PMC4087215.
23: Dufty BM, Warner LR, Hou ST, Jiang SX, Gomez-Isla T, Leenhouts KM, Oxford JT, Feany MB, Masliah E, Rohn TT. Calpain-cleavage of alpha-synuclein: connecting proteolytic processing to disease-linked aggregation. Am J Pathol. 2007 May;170(5):1725-38. PubMed PMID: 17456777; PubMed Central PMCID: PMC1854966.
24: Lewis KA, Su Y, Jou O, Ritchie C, Foong C, Hynan LS, White CL 3rd, Thomas PJ, Hatanpaa KJ. Abnormal neurites containing C-terminally truncated alpha-synuclein are present in Alzheimer’s disease without conventional Lewy body pathology. Am J Pathol. 2010 Dec;177(6):3037-50. doi: 10.2353/ajpath.2010.100552. Epub 2010 Nov 5. PubMed PMID: 21056999; PubMed Central PMCID: PMC2993276.
25: Li W, West N, Colla E, Pletnikova O, Troncoso JC, Marsh L, Dawson TM, Jäkälä P, Hartmann T, Price DL, Lee MK. Aggregation promoting C-terminal truncation of alpha-synuclein is a normal cellular process and is enhanced by the familial Parkinson’s disease-linked mutations. Proc Natl Acad Sci U S A. 2005 Feb 8;102(6):2162-7. Epub 2005 Jan 31. PubMed PMID: 15684072; PubMed Central PMCID: PMC548541.
26: Fleming SM, Ekhator OR, Ghisays V. Assessment of sensorimotor function in mouse models of Parkinson’s disease. J Vis Exp. 2013 Jun 17;(76). doi: 10.3791/50303. PubMed PMID: 23851663; PubMed Central PMCID: PMC3727502.
27: Diepenbroek M, Casadei N, Esmer H, Saido TC, Takano J, Kahle PJ, Nixon RA, Rao MV, Melki R, Pieri L, Helling S, Marcus K, Krueger R, Masliah E, Riess O, Nuber S. Overexpression of the calpain-specific inhibitor calpastatin reduces human alpha-Synuclein processing, aggregation and synaptic impairment in [A30P]αSyn transgenic mice. Hum Mol Genet. 2014 Aug 1;23(15):3975-89. doi: 10.1093/hmg/ddu112. Epub 2014 Mar 11. PubMed PMID: 24619358; PubMed Central PMCID: PMC4110482.
28: Holmqvist S, Chutna O, Bousset L, Aldrin-Kirk P, Li W, Björklund T, Wang ZY, Roybon L, Melki R, Li JY. Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol. 2014 Dec;128(6):805-20. doi: 10.1007/s00401-014-1343-6. Epub 2014 Oct 9. PubMed PMID: 25296989.
29: Recasens A, Dehay B, Bové J, Carballo-Carbajal I, Dovero S, Pérez-Villalba A, Fernagut PO, Blesa J, Parent A, Perier C, Fariñas I, Obeso JA, Bezard E, Vila M. Lewy body extracts from Parkinson disease brains trigger α-synuclein pathology and neurodegeneration in mice and monkeys. Ann Neurol. 2014 Mar;75(3):351-62. doi: 10.1002/ana.24066. Epub 2014 Feb 18. PubMed PMID: 24243558.
30: Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, Lee VM. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012 Nov 16;338(6109):949-53. doi: 10.1126/science.1227157. PubMed PMID: 23161999; PubMed Central PMCID: PMC3552321.
31: Masliah E, Rockenstein E, Adame A, Alford M, Crews L, Hashimoto M, Seubert P, Lee M, Goldstein J, Chilcote T, Games D, Schenk D. Effects of alpha-synuclein immunization in a mouse model of Parkinson’s disease. Neuron. 2005 Jun 16;46(6):857-68. PubMed PMID: 15953415.
32: Kraytsberg Y, Kudryavtseva E, McKee AC, Geula C, Kowall NW, Khrapko K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat Genet. 2006 May;38(5):518-20. Epub 2006 Apr 9. PubMed PMID: 16604072.
33: Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, Jaros E, Hersheson JS, Betts J, Klopstock T, Taylor RW, Turnbull DM. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet. 2006 May;38(5):515-7. Epub 2006 Apr 9. PubMed PMID: 16604074.
34: Chinnery PF. One complex world of mitochondrial parkinsonism. Brain. 2013 Aug;136(Pt 8):2336-9. doi: 10.1093/brain/awt199. PubMed PMID: 23884808; PubMed Central PMCID: PMC3722358.
35:Chinta SJ, Lieu CA, Demaria M, Laberge RM, Campisi J, Andersen JK. Environmental stress, ageing and glial cell senescence: a novel mechanistic link to Parkinson’s disease? J Intern Med. 2013 May;273(5):429-36. doi: 10.1111/joim.12029. Review. PubMed PMID: 23600398; PubMed Central PMCID: PMC3633085.
36:Andersen JK. Senescence and the aging brain. Presentation at Rejuvenation Biotechnology 2014 (RB2014), August 21-23, 2014 Santa Clara, California. Parkinson’s Disease Session. Program p. 41.
37: Arias-Carrión O, Yamada E, Freundlieb N, Djufri M, Maurer L, Hermanns G, Ipach B, Chiu WH, Steiner C, Oertel WH, Höglinger GU. Neurogenesis in substantia nigra of parkinsonian brains? J Neural Transm Suppl. 2009;(73):279-85. Review. PubMed PMID: 20411786.
38: Barker RA, Barrett J, Mason SL, Björklund A. Fetal dopaminergic transplantation trials and the future of neural grafting in Parkinson’s disease. Lancet Neurol. 2013 Jan;12(1):84-91. doi: 10.1016/S1474-4422(12)70295-8. Review. PubMed PMID: 23237903.
39: Loring JF. Negotiating the mine field in the quest for a Parkinson’s disease cell therapy. Presentation at Rejuvenation Biotechnology 2014 (RB2014), August 21-23, 2014 Santa Clara, California. Parkinson’s Disease Session. Program p. 41.
40: Hallett PJ, Cooper O, Sadi D, Robertson H, Mendez I, Isacson O. Long-term health of dopaminergic neuron transplants in Parkinson’s disease patients. Cell Rep. 2014 Jun 26;7(6):1755-61. doi: 10.1016/j.celrep.2014.05.027. Epub 2014 Jun 6. PubMed PMID: 24910427; PubMed Central PMCID: PMC4105701.
41: Kefalopoulou Z, Politis M, Piccini P, Mencacci N, Bhatia K, Jahanshahi M, Widner H, Rehncrona S, Brundin P, Björklund A, Lindvall O, Limousin P, Quinn N, Foltynie T. Long-term clinical outcome of fetal cell transplantation for Parkinson disease: two case reports. JAMA Neurol. 2014 Jan;71(1):83-7. doi: 10.1001/jamaneurol.2013.4749. PubMed PMID: 24217017; PubMed Central PMCID: PMC4235249.
42: Politis M, Wu K, Loane C, Quinn NP, Brooks DJ, Oertel WH, Björklund A, Lindvall O, Piccini P. Serotonin neuron loss and nonmotor symptoms continue in Parkinson’s patients treated with dopamine grafts. Sci Transl Med. 2012 Apr 4;4(128):128ra41. doi: 10.1126/scitranslmed.3003391. PubMed PMID: 22491951.
43: Lindström V, Fagerqvist T, Nordström E, Eriksson F, Lord A, Tucker S, Andersson J, Johannesson M, Schell H, Kahle PJ, Möller C, Gellerfors P, Bergström J, Lannfelt L, Ingelsson M. Immunotherapy targeting α-synuclein protofibrils reduced pathology in (Thy-1)-h[A30P] α-synuclein mice. Neurobiol Dis. 2014 Sep;69:134-43. doi: 10.1016/j.nbd.2014.05.009. Epub 2014 May 20. PubMed PMID: 24851801.
44: Fagerqvist T, Lindström V, Nordström E, Lord A, Tucker SM, Su X, Sahlin C, Kasrayan A, Andersson J, Welander H, Näsström T, Holmquist M, Schell H, Kahle PJ, KalimoH, Möller C, Gellerfors P, Lannfelt L, Bergström J, Ingelsson M. Monoclonal antibodies selective for α-synuclein oligomers/protofibrils recognize brain pathology in Lewy body disorders and α-synuclein transgenic mice with the disease-causing A30P mutation. J Neurochem. 2013 Jul;126(1):131-44. doi: 10.1111/jnc.12175. Epub 2013 Feb 27. PubMed PMID: 23363402.
45: Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest. 2013 Mar 1;123(3):966-72. doi: 10.1172/JCI64098. Epub 2013 Mar 1. Review. PubMed PMID: 23454759; PubMed Central PMCID: PMC3582125.
46: Naylor RM, Baker DJ, van Deursen JM. Senescent cells: a novel therapeutic target for aging and age-related diseases. Clin Pharmacol Ther. 2013 Jan;93(1):105-16. doi: 10.1038/clpt.2012.193. Epub 2012 Dec 5. Review. PubMed PMID: 23212104; PubMed Central PMCID: PMC4051295.
47: Spencer B, Emadi S, Desplats P, Eleuteri S, Michael S, Kosberg K, Shen J, Rockenstein E, Patrick C, Adame A, Gonzalez T, Sierks M, Masliah E. ESCRT-mediated uptake and degradation of brain-targeted α-synuclein single chain antibody attenuates neuronal degeneration in vivo. Mol Ther. 2014 Oct;22(10):1753-67. doi: 10.1038/mt.2014.129. Epub 2014 Jul 10. PubMed PMID: 25008355.