

SENSible Question: What do you think of the investigative report in Science magazine suggesting that the critical study identifying beta-amyloid oligomers as supposedly the key actors in Alzheimer’s disease may have been based on fraudulent data? Wouldn’t that mean that you and other scientists have been wasting all this time chasing a mirage?
Many of the stories in the press that picked up on this investigation sure make it sound that way! Many websites and other media drew readers into their articles using very dramatic headlines, and summarized the findings of the investigation in a way that suggests that this one paper might have single-handedly launched the entire idea of targeting beta-amyloid for removal — and that this paper was likely a fraud. Because of the way the story was structured, even people who read through the original Science investigation too quickly might also have come away with that impression. But as we’ll see, while the investigative report raises grave doubts about the legitimacy of the scientific paper at the heart of the story, the subsequent work of the many other scientists who have worked along superficially similar lines — and, critically, the candidate therapies that have been developed based on that work — are not the tainted fruit of this poisonous tree.
The Science story opens with neuroscientist Matthew Schrag digging into accusations of research fraud not in a basic science study on beta-amyloid, but of malfeasance underlying Simufilam — an experimental Alzheimer’s drug that is currently in clinical trials. The drug company behind Simulafam claims that the drug works by stabilizing a protein in the cell’s system of girders, thereby preventing beta-amyloid from interfering with a receptor involved in memory.
Even before the investor group contracted him to dig into their suspicions, Schrag had tried to replicate some of the critical things that the company’s scientists had reported about the drug, and he’d consistently found that it didn’t work as advertised, so he agrees to look into the investors’ concerns. After documenting what he believes are doctored images in scientific papers and other problems with the underlying science behind Simufilam, Schrag then gets drawn into other cases of alleged scientific malfeasance, and that’s how he comes across the work of former University of Minnesota researcher Sylvain Lesné.
Let’s set the scene for what was happening in the Alzheimer’s field in the early 2000s, when the disputed paper was published. In the 1980s, scientists had identified beta-amyloid (AKA Abeta or Aβ) as the protein responsible for the easiest-to-spot feature of the brains of people who suffer Alzheimer’s-type dementia of aging: the insoluble gloms known as plaques that physically stick to patients’ brain tissue, and to a lesser extent the brains of other aging people. Many scientists spent the ensuing 20 years or so highly focused on this protein as the likely culprit behind the loss of memory and planning and reasoning ability that characterizes neurodegenerative aging of the Alzheimer’s type. But by the early 2000s, more and more researchers had concluded that the idea that the plaques themselves were directly responsible for either cognitive decline or the death of brain cells was on very shaky ground, and they were looking at alternative possibilities.
Most scientists thought that the plaques were too closely tied in with the specific form of dementia diagnosed as Alzheimer’s to have no connection to the disease — but if they weren’t killing neurons or interfering with thinking ability, what was the relationship? Were the plaques merely the wreckage left over after the damage was done? Were they a response to the neurodegenerative changes — maybe even one that might be adaptive? Did they trigger some abnormality in the brain that showed up far away from the plaque itself? Might they even be protective? Or was beta-amyloid indeed the problem after all, but not the kind of beta-amyloid you could see in the brain on autopsy with conventional methods?
The study at the center of the Science investigation provided what looked like compelling evidence to support the last of these possibilities. The paper first noted that mice that produce a mutant form of the precursor protein of beta-amyloid that causes early-onset Alzheimer’s disease in humans begin to show memory deficits long before they develop beta-amyloid plaques or start losing neurons. Several scientific teams had reported this surprising finding several times already by the time the Lesné report came out, but his paper claimed to have fingered the exact molecular culprit behind it. Through several elegant-looking experiments, the article claimed to have winnowed through many different kinds of beta-amyloid that stay dissolved in the fluid surrounding the brain rather than being deposited as plaques, hunting for the one(s) responsible for the pre-plaque loss of memory in these mice. And after seemingly ruling out many of these short “oligomeric” and “proto-fibrillar” forms of aggregated beta-amyloid, Lesné claimed to have finally collared the true villain in the story: a specific beta-amyloid oligomer he called Abeta*56.
This wasn’t a new idea: studies had been trickling in since the 1990s that some form of soluble beta-amyloid was highly disruptive to the brain. For example, the Lesné paper cites two original reports that found this and also two reviews on the subject, including one by his mentor. Additionally, two years before the disputed 2006 Lesné report was published, soluble beta-amyloid aggregates were incriminated by a prominent review article that Lesné did not cite. The review summarized evidence from different labs that implicated short, soluble beta-amyloid assemblies (oligomers and protofibrils) as the real culprits, leaving plaques as mere graveyards or at most reservoirs for these shorter assemblies.
Against that backdrop, Lesné’s 2006 paper seemed to add compelling evidence in support of the soluble-species hypothesis. It therefore sent many researchers into their labs with a new focus on these species, performing experiments to further test the idea and looking for ways to block or remove oligomers and protofibrils from the brains of mice (and, they hoped, eventually people suffering with Alzheimer’s).
Years later, looking at the images in Lesné’s published work on beta-amyloid, Schrag saw multiple red flags. And when Science magazine asked scientific image analysts and prominent Alzheimer’s researchers to look at the suspect figures, they too saw problems with literally hundreds of Lesné’s published images — starting with the 2006 paper and extending to more than 70 of his other scientific reports, including what one researcher described as some “shockingly blatant” cases of apparent image tampering.
With that setup, it’s easy to fall into thinking that Lesné’s suspect research is the Original Sin behind the many failed Alzheimer’s drug candidates that have littered the scientific landscape like so many of the jury-rigged vehicles strewn across the desert at the end of a Mad Max movie. But in fact, as the investigative report itself notes, nearly no one else in the field outside of Lesné’s own collaborators has worked with the specific beta-amyloid oligomer (Abeta*56) he had fingered in his reports!
Yes, the 2006 paper was widely read, trumpeted, and cited. Yes, it refocused the field on the general idea that the soluble forms of beta-amyloid rather than the plaques were the root drivers of Alzheimer’s-type neurodegenerative aging. But no: the rest of the field has not spent the last 16 years tilting their pipettes at the molecular windmills of Abeta*56 —because they couldn’t replicate his methods or findings.

The Science investigation itself mentions this critical fact here and there throughout the article — but casually, and without spelling out how it affected how the Lesné paper did — and did not — shape Alzheimer’s research in the ensuing decade and a half.
For instance, the Science piece reports that “In the 16 years following the landmark paper, Lesné and Ashe—separately or jointly—published many articles on their stellar oligomer. Yet only a handful of other groups have [even] reported detecting Aβ*56 [my emphasis].” Other points in the report that seem to have gotten similarly lost in the drama of accusations of skullduggery bear quoting in full (all emphasis is mine):
even before Schrag’s investigation, the spotty evidence that Aβ*56 plays a role in Alzheimer’s had raised eyebrows. [Alzheimer’s expert Donna] Wilcock has long doubted studies that claim to use “purified” Aβ*56. Such oligomers are notoriously unstable, converting to other oligomer types spontaneously. Multiple types can be present in a sample even after purification efforts, making it hard to say any cognitive effects are due to Aβ*56 alone, she notes—assuming it exists.
In fact, Wilcock and others say, several labs have tried and failed to find Aβ*56, although few have published those [negative] findings. Journals are often uninterested in negative results, and researchers can be reluctant to contradict a famous investigator.
An exception was Harvard University’s Dennis Selkoe, a leading advocate of the amyloid and toxic oligomer hypotheses, who has cited the [2006] Nature paper at least 13 times. In two 2008 papers, Selkoe reported that he could not find Aβ*56 in human fluids or tissues.
Dr. Selkoe is indeed “a leading advocate of the amyloid and toxic oligomer hypotheses.” He is perhaps the most prominent and vocal voice in the scientific community in formulating and defending the contemporary “Amyloid Cascade” hypothesis, which has beta-amyloid oligomers and protofibrils at its center. In fact, years before the appearance of the disputed 2006 Lesné paper, he was one of several researchers who had already zeroed in on beta-amyloid oligomers as the more likely culprit in Alzheimer’s-type neurodegenerative aging, and he had published an influential review laying out the evidence two years earlier.
So Selkoe was doubtless initially gratified when one of the most reputable scientific journals published a strong-looking scientific paper that seemed to validate the hypothesis he had come to accept. But as a scientist, he didn’t just blindly accept Lesné’s report, even if it supported his understanding of the disease process. He looked for Aβ*56 in human brain tissue and the fluid surrounding the brain — and when he couldn’t find it, he didn’t just scratch his head and move on to the next thing, but reported its absence in published scientific papers.
But a no-show for Aβ*56 didn’t cause him to abandon the central idea that soluble forms of beta-amyloid are the core driver of the disease, because his own findings and those of many other scientists repeatedly found evidence that supported the model— completely independently of Lesné and Abeta*56.
You Won’t Catch Tropical Fish in the Arctic
And here’s the critical point. Since virtually no one else even “saw” Aβ*56 in their studies, let alone thought Aβ*56 was a central player in Alzheimer’s, no scientist developed a therapy intended to target it. So the paper has hardly sent the scientific community chasing after gremlins for the last 16 years.
The Science investigation either fails to spell this out clearly, or intentionally ignores it, or was simply too focused on the narrow questions around Lesné’s publication history to place it in its proper context. In one oddly-constructed sentence, the author writes: “Like other anti-Aβ efforts, toxic oligomer research has spawned no effective therapies.” That was strictly speaking true at the time the Science investigation was published — writing it that way might lead the reader to mistakenly assume that the “toxic oligomer research ha[d] spawned” dozens of anti-oligomer candidates, and none of them had proved effective. Instead, the lack of a crop of therapies targeting soluble beta-amyloid aggregates results from nearly no such treatments having ever been developed. In particular, no biotech company or Big Pharma player has ever designed an antibody to target Aβ*56. And more broadly, they’ve also developed next to none that effectively target any of the other protofibrils or oligomer species that Selkoe and others have implicated.
This is a critical point — about this study and the path to ending the misery and suffering of neurodegenerative aging. When people express skepticism that beta-amyloid is driving neurodegenerative aging of the Alzheimer’s type, they often point to the many failed therapies that have tried to reduce the level of beta-amyloid in the brain one way or another. But in doing so, critics tend to throw many different kinds of therapies into a kind of mental food processor. The result is a kind of cerebral junk food, glowing with an artificial “health halo” thanks to its inclusion of a few healthy high-value ingredients but mostly comprised of low-value filler material. It’s no surprise if you wind up intellectually ill-nourished if you make such a mishmash the centerpiece of your intellectual diet.
Instead, if we really want to understand what’s been going on for the last twenty years, we have to look in detail at how individual therapies are supposed to work; how that lines up with our understanding of beta-amyloid’s relationship to dementia; and whether any individual treatment actually does what its designers intended it to do in the brain if it is to help clear the dark cobwebs from the mind.
To understand what’s actually been holding us back in recent years, let’s start by disentangling (pun intended) the different forms that beta-amyloid can take in the brain. Brain nerve cells produce beta-amyloid in the form of individual protein molecules called monomers. Monomers of beta-amyloid are already bent out of the normal conformation of the underlying protein, which makes them prone to act as a kind of malevolent Lego®, with each monomer easily snapping into place in the slot provided by a previous monomer, allowing them to form larger and larger chains. At the very far end of this process, the insoluble plaques that are the easiest-to-spot form of beta-amyloid seen in the brains of people who die suffering with Alzheimer’s are huge sepulchers made up of long, twisted fibrils of these molecular Lego®s.
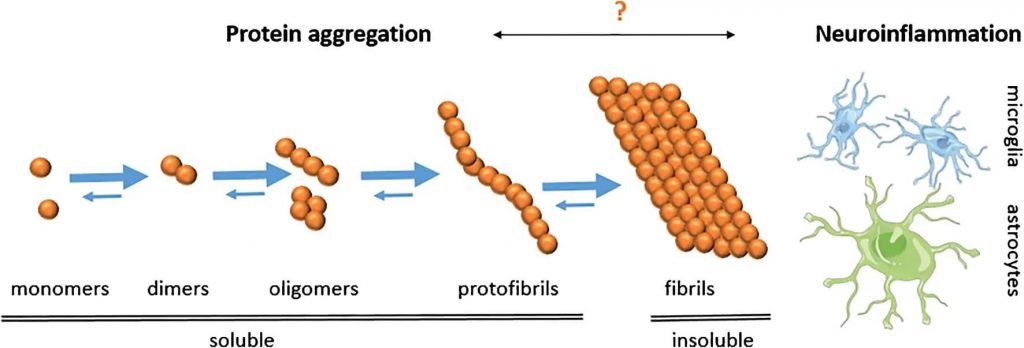
But in the early stages of this process, when only a few beta-amyloid monomers have snapped together, the very short chains they form remain dissolved in the fluid surrounding the brain instead of forming plaques. These include oligomers and the slightly longer protofibrils that most scientists today blame for directly disrupting brain neuron function and for indirectly setting off the cascade that leads to the other characteristic damage in the Alzheimer’s patient brain (abnormal forms of the protein tau and the loss of specific populations of neurons). Because of their solubility, these shorter beta-amyloid assemblies are more challenging to detect and study, and the very methods used to extract them can cause them to stick together and form larger assemblies. Therefore, unless researchers take great care, the form of beta-amyloid they wind up studying may be a different, larger assembly than the form that was originally present in a mouse or human brain. As we’ve noted, many of Dr. Lesné’s colleagues think he may in part have fallen into this trap.
So when someone blithely says that all the drugs targeting beta-amyloid have failed, what kinds of drugs are we talking about?
To begin with, many of the failed drugs targeting beta-amyloid have tried to protect the brain by interfering with brain neurons’ ability to produce beta-amyloid in the first place. Part of SRF founder Dr. Aubrey de Grey’s central motivation for developing the SENS “damage-repair” approach was his recognition of the futility of applying this kind of conventional “messing-with-metabolism” strategy to neurodegenerative aging or other so-called “diseases” of aging. And in this case, drugs whose mechanism involved inhibiting beta-amyloid biosynthesis didn’t just fail to make patients better: these drugs — including verubecestat, lanabecestat, atabecestat, and most famously Eli Lilly’s semagacestat — actually made cognitive function even worse!
It’s not entirely clear why these drugs had the opposite of their intended effect, but it’s bound to be broadly tied up with the basic fact that the regulated biological systems they target evolved for a reason — and the reason is not that natural selection has favored the retention of genes that lead us into age-related dementia and death. One of the more likely explanations for their failure is that the enzymes in the brain that these drugs inhibit have other essential targets in the brain and elsewhere, so the drugs’ toxicity may be due to their unintended interference with these other functions. Alternatively, these drugs might cause problems in the aging brain because beta-amyloid itself serves a short-term biological purpose, such as neutralizing pathogens in the brain, so blocking its synthesis leaves the brain even more vulnerable to these microbes’ attack.
Regardless of the specific reason, the disastrous failure of these drugs does not exonerate beta-amyloid as the central actor behind neurodegenerative aging of the Alzheimer’s type. Instead, their failure is an object lesson in the general principle that he who grasps at the rails of metabolism is likely to be electrocuted.
Even when we turn to the SENS strategy of removing existing aging damage of aging directly, we still see a lot of failures — but once again, the reasons why become clearer when we dig into the details. We’ll first note that literally no treatment targeting beta-amyloid that reached clinical trials was designed to remove Lesné’s elusive (or illusive?) Aβ*56. So it’s not as if any potential scientific misconduct embedded in that paper has dragged the field into creating a slough of failed Aβ*56-targeting therapies.
And while it may be a good thing that no one was actually putting patients at risk to test an Aβ*56-targeting therapy that (it appears) would never have had a chance of success, it’s also true that all of the Alzheimer’s therapies that were tried and didn’t work failed to meaningfully engage any form of soluble beta-amyloid at all!
Bapineuzumab, which was one of the first antibody-based AmyloSENS candidates to fail in clinical trials, mostly targeted the plaques themselves. Similarly, gantenerumab (which failed and was resurrected, only to fail again) was specifically chosen for its strong binding to full-length beta-amyloid fibrils. At the other end of the spectrum, Eli Lilly’s solanezumab didn’t target oligomers or protofibrils either: instead, it targeted monomers that hadn’t formed any kind of Lego® assembly at all. Whatever benefit that might have, it might be counteracted by interfering with the possible antimicrobial host defense activity of beta-amyloid in the brain. And there’s also evidence that solanezumab might not have actually removed beta-amyloid from the brain at all, but instead captured it, recirculated it, and then dropped it off right where it came from!

And while scientists intended crenezumab to target oligomers, when actually tested, its activity turned out to be all over the place. Yes, it did bind some oligomeric forms of beta-amyloid as intended — but it also bound to full-on fibrils and even some monomers, suggesting that the infused antibodies might have been “spread too thin” across different forms of beta-amyloid make a meaningful dent in the brain’s burden of toxic soluble beta-amyloid species.
In fact, only one of the AmyloSENS candidates that have reached advanced clinical testing in humans has effectively removed short, soluble beta-amyloid assemblies from the brain: lecanemab. In a recent head-to-head comparison, lecanemab locked onto protofibrils ten times more strongly than to mature fibrils. That made lecanemab a much more selective therapy for protofibrils than other tested antibodies: gantenerumab bound very small oligomers well but protofibrils only weakly, and the now-controversial aducanumab (Aduhelm®) bound modestly. Instead, both gantenerumab and aducanumab bound more strongly to full-on fibrils, which lecanemab barely touched. And lecanemab and aducanumab bind only very weakly to monomers.
For this exact reason, even self-described “amyloid skeptics” pointed to lecanemab as “one we had a lot of hope for” from the early days of its development. And as you’ve likely heard, protofibril-targeting lecanemab has finally bent the curve on neurodegenerative aging of the Alzheimer’s type, slowing the disease’s terrible downward spiral in its early clinical stages by 27%. While some people are now pooh-pooing this result, it’s actually a remarkable breakthrough: not only is it the first therapy in history to unambiguously put the brakes on the runaway train that is age-related neurodegeneration, but it did so in the face of having the deck very much stacked against it, for reasons we’ll discuss in a future post.
Summing Up — and Moving On
So by pointing the accusing finger squarely at beta-amyloid oligomers, the Lesné paper certainly had an important role in attracting scientists to the modern version of the “Amyloid cascade.” To that extent, it has turned out ironically to be a pivotal paper in getting us to where we are today.
But that’s about where its influence ended. Nearly no one else in the field got waylaid by chasing after Aβ*56; no biotech VC or pharma giants wasted huge chunks of its R&D budget on therapies intended to target it; no patients were put at risk in clinical trials testing therapies that were in any way intended to counteract its supposedly insidious effects on the brain. And when the first candidate AmyloSENS therapy to finally target soluble beta-amyloid species in the brain reached Phase III clinical trials, it worked, acting like a parachute for people suffering neurodegenerative aging.
To save the precious human mind from the ravages of neurodegenerative aging, we will need more than parachutes — but parachutes we do need. That’s the nature of SENS: it’s a “divide-and-conquer” approach. Clearing beta-amyloid out of the brain is one crucial step toward keeping the brain youthfully sharp and attentive, but we will also need to remove aberrant tau inside neurons and sustain neuronal circuits with neuronal replacement therapies — both projects under active pursuit by SENS Research Foundation scientists and SRF-funded external researchers. Once we have all three, and especially once we are able to comprehensively clear out the full scope of the cellular and molecular damage of aging, we will fully engage our cognitive jetpacks — and soar.
As with achieving Longevity Escape Velocity, arresting and reversing neurodegenerative aging depends on how much damage a person has already suffered when treatment begins and the comprehensiveness, safety, and effectiveness of the suite of rejuvenation biotechnologies applied. Removing beta-amyloid protofibrils alone slows the rate of decline, but cannot arrest or reverse it without additional damage-removal strategies. Credit: ADNJ de Grey, PLoS Biol 2(6):723-6.