
Aggregates of beta-amyloid (Aß) and other malformed proteins accumulate in brain aging and neurodegenerative disease, leading progressively to neuronal dysfunction and/or loss. The regenerative engineering solution to these insults is therapeutic clearance of aggregates, extracellular (such as Aß plaques) and intracellular (such as soluble, oligomeric Aß). Immunotherapeutic Aß clearance from the brain is a very active field of Alzheimer’s research, with at least seven passive, and several second-generation active, Aß vaccines currently in human clinical trials.(1)
One challenge to optimal vaccine design is matching the specificity of antibodies to the range of Aß aggregates that form in vivo, including different oligomeric and protofibrillar assemblies in addition to the Aß fibrils that compose the hallmark plaques of aging Alzheimer’s disease (AD) brain. Research has shown that agents that sequester one Aß species may leave other species intact, and in some cases a shift in assembly dynamics can actually promote the formation of one species while clearing or reducing the formation of others (eg, (2-6)). On the other hand, insufficiently Aß-specific antibodies may target the physiological amyloid precursor protein (APP). While the balance of evidence suggests that toxic oligomeric species are the most impairing Aß assembly, and while some researchers even believe that the insoluble plaques may act as protective “sinks” that sequester more toxic Aß species, all such aggregates are stochastic accretions to the aging brain, and the ideal Aß-targeting strategy would lead to the clearance of all Aß aggregates while not intervinging with APP metabolism.
Although in very early in vivo testing, a new approach has emerged that may offer that promise. This is the use of an Aß-targeting affibody, ie, a novel non-immunoglobulin binding protein generated through combinatorial protein engineering, using phage display to select ligands from a non-cysteine three-helix bundle domain library.(7) In 2007, a team from Sweden’s Royal Institute of Technology (KTH) reported the generation of the selective affibody ZAβ3, which forms a disulfide-linked dimer that binds with nanomolar affinity to Aß monomers.(8) Upon binding to Aß, the ZAβ3 dimer forms stable complexes with Aß, forcing it into a hairpin conformation and safely burying its hydrophobic, aggregation-prone domains within its structure (Fig 1).
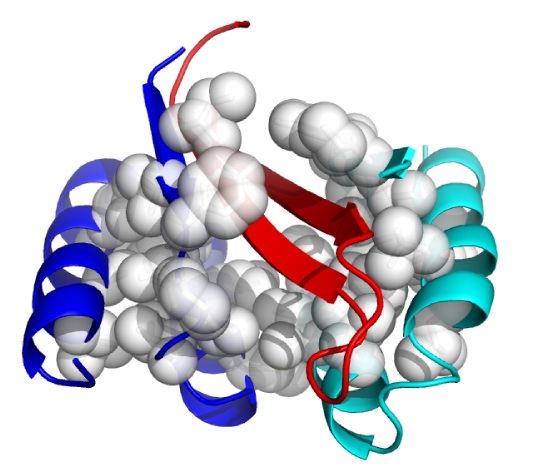
Figure 1: ZAβ3 (blue and cyan; nonpolar side chains in white) complexed to an Aβ40 hairpin at residues 16-40 (red). From (9).
To test the effects of ZAβ3 on Aß aggregatess and neurotoxicity in vivo, Swedish and English collaborators used Drosophila lines transgenically expressing either wild-type Aß42, E22G (the preferrentially oligomer-forming “Arctic” Aß mutation first identified in a Swedish familial AD proband), or the relatively benign, non-aggregative isoform Aß40. These were then crossed with strains transgenic for ZAβ3; for 2 copies of ZAβ3 connected head-to-tail ((ZAβ3)2) to facilitate formation of the ZAβ3 dimer; or for overexpression of the wild-type Z domain alone (as controls). The resulting hybrids were crossed onto strains bearing drivers to ensure coexpression of the transgenes in brain neurons or photoreceptors.(9)
Transgenic Aß42 is severely neurotoxic to Drosophila, reducing lifespan from 38 days in controls to 28 d (WT Aß42) or 9 d (E22G). E22G neurotoxicity was also evident in the eye, where expression led to abnormal photoreceptor morphology. ZAβ3 substantially rescued fly lifespan against the expressed Aß, to 32 and 20 d, respectively. (ZAβ3)2 proved yet more effective: it fully rescued lifespan from the effects of WT Aß42 (to 40 d) and more fully normalized it against the effects of E22G (31 d), while substantially protecting photoreceptor morphology. Aß40 was harmless to the flies, and lifespan of TG-Aß40 Drosophila was unaffected by coexpression of affibodies.
Affibody clearance of Aß E22G was also evident, with Aß levels being high in E22G-TG Drosophila but considerably reduced in flies coexpressing ZAβ3 and indetectable with coexpression of (ZAβ3)2. That the mechanism of Aß reduction was not due to effects on Aß production was demonstrated by the lack of a significant effect on neuronal E22G expression. More intriguingly, the reduction in Aß was also not accompanied by a concomitant buildup in SDS-stable ZAβ3-Aß complexes, as demonstrated by a lack of appearance of liberated Aß following treatment with guanidinium chloride to dissociate such complexes. Rather, the absolute, total level of Aß in E22G-TG Drosophila was ~75% lower in flies coexpressing ZAβ3 monomer, and by 97±3% with coexpressed (ZAβ3)2, relative to levels in controls coexpressing inert Z domain.(9)
Reductions in monomeric Aß were accompanied by equivalent reductions in Aß fibrils in vivo, and in vitro studies showed that addition of affibody to Aß42 or -40 isoforms exerts equivalent effects on the kinetics of fibril formation as do equivalent reductions in initial levels of Aß monomer in the medium, implying that the reduction in fibril levels is primarily due to the initial binding of affibody to Aß monomer. Yet addition of excessive affibody later in the aggregation reaction process also arrests further fibril formation and even leads to a very gradual reduction in levels of existing fibrils, accompanied by the appearance of affibody-bound Aß monomers; this result suggests some ability to bind Aß monomers within fibrillar and protofibrillar Aß structures, and to slowly contribute to their dissociation (although at rates so low as to be unlikely to occur in situ in the brain).(9)
On the other hand, similar studies not only revealed a similar ability of ZAβ3 to prevent the formation of Aß oligomers from monomeric Aß, but also a physiologically plausible destabilization and dissolution of existing oligomers.(9)
The sum of the results suggest that initial sequestration of Aß monomers by ZAβ3 — as free Aß molecules or as constituents of oligomeric and perhaps protofibrillar or even fibrillar Aß — is later followed by degradation of affibody-bound Aß in vivo. The authors note that the mechanism underlying any such effect cannot involve clearance of Aß species by anti-Aß antibodies induced in the flies through activation of an adaptive immune response, as Drosophila do not have an adaptive immune system. Rather, they suggest that affibody-bound Aß monomers are subsequently degraded by endogenous biological mechanisms, such as proteasomal or lysosomal breakdown within neurons, or by phagocyes following an hypothetical exocytosis.(9) If correct, the rate of such degradation could in principle be enhanced if necessary by fortification of the lysosome with novel hydrolases (lysoSENS) targeting residual lysosomal Aß accumulations or any specific degradation-resistant aggregates formed following lysosomal uptake and storage.(13-15)
Obviously, these studies, while promising, are very preliminary. Drosophila is not an ideal model organism in which to test the effects of Aß and its clearance in vivo, and neuronal ZAβ33 coexpression studies can only be taken as suggestive of the possible effects of affibody delivered by systemic injection. Moreover, there remain many open questions and concerns about the timecourse and mechanism of Aß disappearance, such as whether ZAβ33 can indeed clear out existing accumulations of Aß oligomers and fibrils in aging human brains, and whether a mechanism apparently largely reliant on sequestration of free monomers might interfere with any putative physiologic role of Aß in the brain(10-12).
But a novel route toward Aß removal that does not rely on the immune system, and that offers the promise of removal of a wide range of Aß aggregates but especially of oligomeric species, is a promising new development in the field. Indeed, while speculation from this preliminary result cannot be given much weight, under a best-case scenario one can envision the opening of an entirely new approach to therapeutic clearance not only of Aß, but of a range of other disease- and aging-associated intracellular and extracellular aggregates. The broad outlines of the line of investigation required to turn these preliminary results into a therapy for brain aging and AD is reasonably clear; if its potential is realized clinically, then affibody-based therapeutics may ultimately be used to prevent and even reverse age-related diseases ranging from other forms of neurodegeneration, to senile cardiac amyloidosis, to age-related macular degeneration, to atherosclerosis. We await further revelations from these investigators with tempered optimism.
References
1. Lemere CA, Masliah E. Can Alzheimer disease be prevented by amyloid-beta immunotherapy? Nat Rev Neurol. 2010 Feb;6(2):108-19. Review. Erratum in: Nat Rev Neurol. 2010 Apr;6(4):183. PubMed PMID: 20140000; PubMed Central PMCID: PMC2864089.
2. Mamikonyan G, Necula M, Mkrtichyan M, Ghochikyan A, Petrushina I, Movsesyan N, Mina E, Kiyatkin A, Glabe CG, Cribbs DH, Agadjanyan MG. Anti-A beta 1-11 antibody binds to different beta-amyloid species, inhibits fibril formation, and disaggregates preformed fibrils but not the most toxic oligomers. J Biol Chem. 2007 Aug 3;282(31):22376-86. Epub 2007 Jun 1. PubMed PMID: 17545160; PubMed Central PMCID: PMC2435219.
3. Necula M, Breydo L, Milton S, Kayed R, van der Veer WE, Tone P, Glabe CG. Methylene blue inhibits amyloid Aß oligomerization by promoting fibrillization. Biochemistry. 2007 Jul 31;46(30):8850-60. Epub 2007 Jun 27. PubMed PMID: 17595112.
4. Necula M, Kayed R, Milton S, Glabe CG. Small molecule inhibitors of aggregation indicate that amyloid beta oligomerization and fibrillization pathways are independent and distinct. J Biol Chem. 2007 Apr 6;282(14):10311-24. Epub 2007 Feb 6. PubMed PMID: 17284452.
5. Wang MS, Boddapati S, Sierks MR. Antifibrillizing agents catalyze the formation of unstable intermediate aggregates of beta-amyloid. Biotechnol Prog. 2010 Feb 8. [Epub ahead of print] PubMed PMID: 20306540.
6. Petrushina I, Ghochikyan A, Mktrichyan M, Mamikonyan G, Movsesyan N, Davtyan H, Patel A, Head E, Cribbs DH, Agadjanyan MG. Alzheimer’s disease peptide epitope vaccine reduces insoluble but not soluble/oligomeric Aß species in amyloid precursor protein transgenic mice. J Neurosci. 2007 Nov 14;27(46):12721-31. PubMed PMID: 18003852; PubMed Central PMCID: PMC2366938.
7. Nygren PA. Alternative binding proteins: affibody binding proteins developed from a small three-helix bundle scaffold. FEBS J. 2008 Jun;275(11):2668-76. Epub 2008 Apr 24. Review. PubMed PMID: 18435759.
8. Grönwall C, Jonsson A, Lindström S, Gunneriusson E, Ståhl S, Herne N. Selection and characterization of Affibody ligands binding to Alzheimer amyloid beta peptides. J Biotechnol. 2007 Jan 30;128(1):162-83. Epub 2006 Sep 27. PubMed PMID: 17088007.
9. Luheshi LM, Hoyer W, de Barros TP, van Dijk Härd I, Brorsson AC, Macao B, Persson C, Crowther DC, Lomas DA, Ståhl S, Dobson CM, Härd T. Sequestration of the Aß peptide prevents toxicity and promotes degradation in vivo. PLoS Biol. 2010 Mar 16;8(3):e1000334. PubMed PMID: 20305716; PubMed Central PMCID: PMC2838747.
10. Morley JE, Farr SA, Banks WA, Johnson SN, Yamada KA, Xu L. A physiological role for amyloid-beta protein: enhancement of learning and memory. J Alzheimers Dis. 2010 Jan;19(2):441-9. PubMed PMID: 19749407.
11. Abramov E, Dolev I, Fogel H, Ciccotosto GD, Ruff E, Slutsky I. Amyloid-beta as a positive endogenous regulator of release probability at hippocampal synapses. Nat Neurosci. 2009 Dec;12(12):1567-76. PubMed PMID: 19935655.
12. Pearson HA, Peers C. Physiological roles for amyloid beta peptides. J Physiol. 2006 Aug 15;575(Pt 1):5-10. Epub 2006 Jun 29. Review. PubMed PMID: 16809372; PubMed Central PMCID: PMC1819417.
13. de Grey AD, Alvarez PJ, Brady RO, Cuervo AM, Jerome WG, McCarty PL, Nixon RA, Rittmann BE, Sparrow JR. Medical bioremediation: prospects for the application of microbial catabolic diversity to aging and several major age-related diseases. Ageing Res Rev. 2005 Aug;4(3):315-38. PubMed: 16040282.
14. de Grey AD. Appropriating microbial catabolism: a proposal to treat and prevent neurodegeneration. Neurobiol Aging 2006;27(4):589-595. PubMed: 16207503.
15. de Grey AD. Lysosomal enhancement with microbial hydrolases: a novel strategy for removing protein aggregates. In: A. Fisher et al. (eds), New Trends in Alzheimer and Parkinson Disorders: ADPD 2005. Medimond, 2005: 51-4.