

New Human Trial Revelations Show Clearing Beta-Amyloid Early is Even More Effective
Short summary: The Phase III trials for AmyloSENS rejuvenation biotechnologies lecanemab/Leqembi® and donanemab showed that they are most effective when given to people with less of other kinds of cellular and molecular aging damage in their brains. New data illustrates that fact even more powerfully and gives us a foreshadowing of what’s possible if we make best use of these and forthcoming damage-repair longevity therapeutics.
In a previous blog post, we shared the thrilling news that clinical trials had proven that the AmyloSENS therapies donanemab and lecanemab (Leqembi®) slow the terrible fall into neurodegenerative aging of the Alzheimer’s type (AD). As we noted, one key reason these trials succeeded where many promising antibodies had failed is that they started giving people these treatments earlier in the course of the disease. The reason why early treatment is critical is not primarily because there’s less beta-amyloid in the brain earlier on in the course of AD. Although that would be true if they had started these trials in people who were still cognitively intact, these trials included only volunteers who were already at a stage in which the level of plaque in the brain has generally reached a plateau (though it’s less clear what happens to the level of the smaller assemblies of beta-amyloid – i.e., the oligomers and protofibrils — that remain dissolved in brain fluid).
Instead, the benefit of acting early comes from the greater opportunity for beta-amyloid clearance to hold off other kinds of aging damage that occur downstream of it in the brain. At the 16th Clinical Trials on Alzheimer’s Disease conference (CTAD), the companies behind both of these powerful medicines revealed more data that reinforces and extends this vital lesson. Some of the revelations come from additional analysis of the Phase III trials; others emerged from further followup on trial participants; and still more came out of a separate trial whose first results the companies made public for the first time.
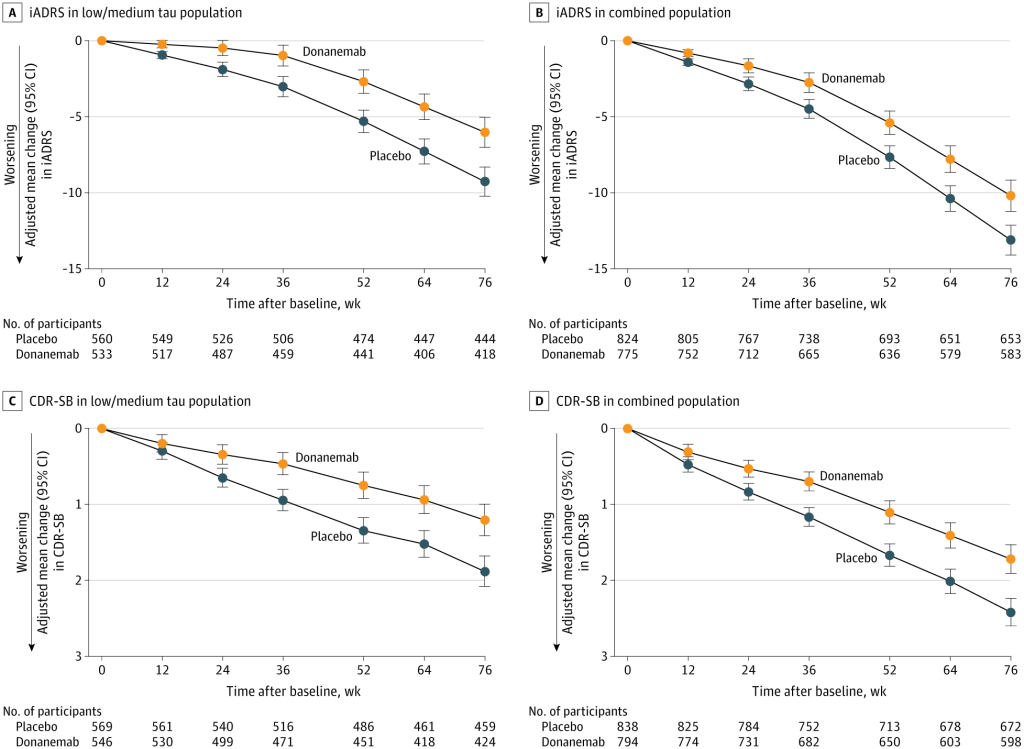
In the original Phase III trial for donanemab, the researchers didn’t just compare all the subjects who received the antibody to those who received the placebo, but also compared people who received the treatment and who had “moderate” levels of tau aggregates in their brains to all the treated subjects combined (that is, those with moderate levels plus those with high levels together).*
As we discussed in the previous blog post, donanemab slowed the downward drag of the disease in all groups, but it was more effective in people who were less burdened by brain tau aggregates. When scientists used the integrated Alzheimer Disease Rating Scale (iADRS) to test donanemab’s effectiveness in preserving the ability of people in the trial to carry on the day-to-day business of life and social interaction, they found that it slowed the fall by 35% in people with a medium tau burden, but by only 22% in the combined population. And when they tested cognitive function more narrowly using the Clinical Dementia Rating – Sum of Boxes (CDR-SB) score, donanemab treatment preserved 36% of the cognitive function in the medium-tau population, but just 29% in the combined population.
And this superior slowing emerged despite a built-in handicap in the combined population score. The researchers compared the medium-tau volunteers who were treated with donanemab to a matching placebo group who also had moderate tau aggregates in their brains. But this placebo group declined more slowly in the first place than the placebo group for the combined population (see the Figure). So on its face, there was less room for the antibody to improve the medium-tau group’s trajectory.
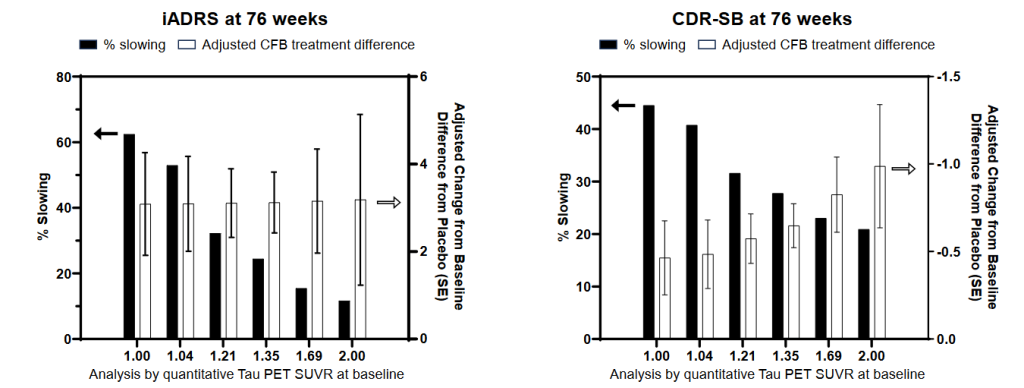
In a presentation at CTAD, scientists from the trial team presented a more granular analysis, ranking the patients into six groups ranging from those with the lowest burden of tau aggregates in their brains to those with the highest. This stronger gradient showed even more clearly the benefit of clearing beta-amyloid before it drives too much progression of aberrant tau. While the antibody benefited people across the full range of tau aggregate scores, donanemab had the clearest effect in people who started the trial with the lowest burden of tau aggregates in their brains (SUVR 1). In such low-tau aggregate people, donanemab slowed functional and cognitive decline by a remarkable 60% on the iADRS and 45% on the CDR-SB!
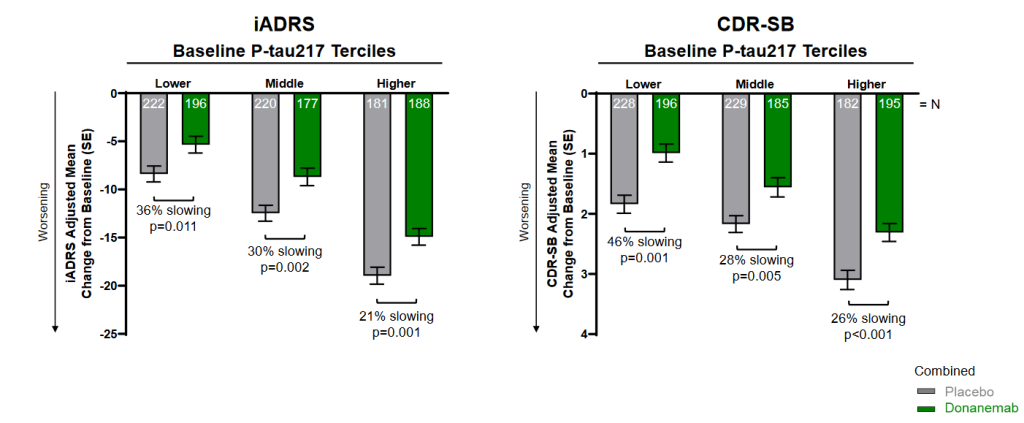
Donanemab was similarly more effective in people with a lower baseline burden of other signs and forms of aging damage. For example, the researchers compared the AmyoSENS therapy’s effect in people with lower or higher levels of ptau-217, a specific form of abnormal tau that can be picked up in the plasma. Researchers have found that plasma ptau-217 closely reflects the burden of both beta-amyloid and tau in the brain. Accordingly, levels of ptau-217 predict the onset of AD and rise as AD worsens, whereas it does not stand out in people suffering from other neurodegenerative diseases. Even when subjects received placebo infusions, having a lower level of ptau-217 when the trial began was associated with less loss of cognitive function by the end of the trial. And in people who instead received donanemab, lower initial ptau-217 made the antibody even more effective at braking the disease course.
When the investigators then layered a person’s calendar age onto his or her plasma ptau-217 level, they saw that having lower levels of the tau-based biomarker predicted greater resilience against cognitive decline in both younger and older volunteers — but that even at similar levels of tau damage marker, younger volunteers benefitted even more from donanemab than older volunteers did. This is as you would predict, because although people of similar ages do vary somewhat in their biological age, calendar age itself is still a highly reliable metric for “all-in” aging damage. So in this analysis, calendar age acts as a proxy for a wide range of cellular and molecular aging damage impacting the volunteers’ brains, such as having suffered “silent strokes,” damage to the small blood vessels and the insulating material for the “wiring” in the brain, senescent cells, mitochondrial mutations, and all the other carnage that aging imposes on our brains.

Of course, any individual person may be moderately biologically younger or older (that is, have a somewhat lower or higher burden of aging damage) than the average person of the same chronological (calendar) age. More subtly, a given person may have age-typical levels of most kinds of aging damage but have especially high burdens of damage in specific organs or systems, which is why some people develop particular diseases of aging earlier than others.
What we see in this graph is a case in point. There were people in both the older and the younger subgroup who had greater or lower ptau-217 levels. But by further dividing the participants into younger and older subgroups, the scientists revealed the effects of the other, less AD-specific damage that was also afflicting the older volunteers’ brains.
And on top of all that, early donanemab treatment yielded a significantly greater reduction in the number of cells called astrocytes in the brain that had abandoned their normal housekeeping activities and become “reactive.” When the brain is under attack in AD and other conditions, astrocytes swell up and try to defend the brain from attack by swallowing up and processing beta-amyloid, and also by forming what is called the “glial scar” — a barrier that is intended to wall vulnerable neurons off from pathogens and other noxious agents. Unfortunately, these “reactive astrocytes” are highly inflammatory, which can damage the brain and derange its signaling — and the protective glial scar can act like the “peace walls” in Northern Ireland, forming a barrier that prevents new neural growth from reinvigorating damaged circuits.
As you would expect from the fact that the trigger for reactive astrocytes in the AD-prone brain is the high levels of beta-amyloid, clearing out amyloid using donanemab allowed reactive astrocytes to calm down again, shrinking many of them back to their normal size. People in the low-damage “addendum” group had lower levels of these inflammatory cells when the study began — and donanemab lowered their numbers even further, leaving them with the fewest reactive astrocytes of any group.
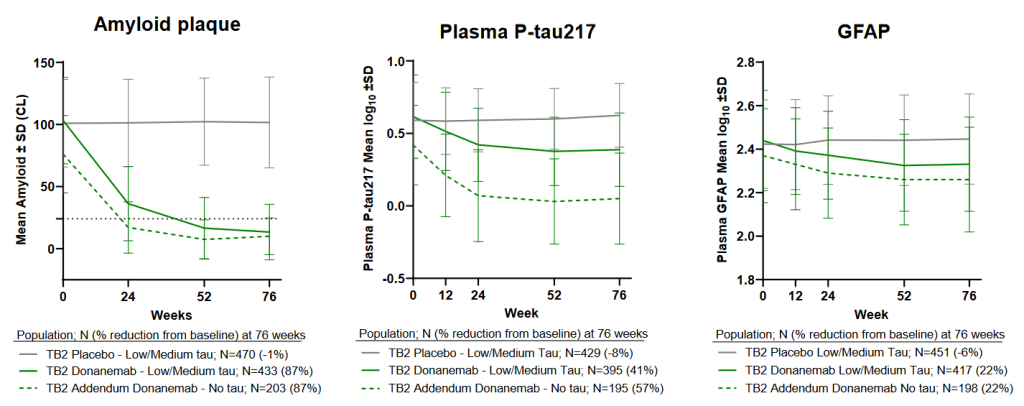
And in a separate presentation, Lilly scientists showed that even though all patients were treated with donanemab until they got their brain beta-amyloid levels down to a low goal level, speed mattered. The faster a person reached their beta-amyloid clearance goal, the more effective donanemab was at slowing cognitive function and the less they would suffer additional tau aggregate accumulation in their brains.
Getting a Head Start
In addition to their deeper dive into the donanemab Phase III trial, Lilly researchers teased the very beginnings of what would have to be a much longer study. Would-be participants in the Phase III trial for donanemab had to have at least what they call “moderate” levels of tau aggregates in their brains (a score of 1.10 to 1.46 SUVR on PET scans in the graph above) to qualify. This requirement might seem perverse in retrospect, since the trial results show that people with lower levels of tau aggregates benefit more from donanemab, and scientists have predicted for a decade or more that starting even earlier could head off the clinical disease entirely. But the flip side is that if Lilly had launched a trial that included people with only minimal levels of AD-related molecular damage, it would have taken much longer for people receiving the placebo to suffer obvious declines in their memory and self-governance — and thus for donanemab to show that it could slow or prevent that decline. Lilly therefore chose to run the trial in people with higher amounts of damage to get decisive results quickly.
But while people with low levels of beta-amyloid and next to no tau damage in their brains could not sign up for the trial itself, Lilly offered some of them the opportunity to participate in what they called an “addendum” study. All the participants in the addendum would be guaranteed to get donanemab, and Lilly scientists would track their progress; there would be no placebo group. A little over a thousand people signed up.
Donanemab cleared beta-amyloid out of the brains of these “early-adopter” patients at the same pace that it had in people with more damage. But because they began the study with a lower level of beta-amyloid even before receiving their first infusion, these very-low-damage volunteers reached maximum clearance more quickly than those in the main trial. Additionally, donanemab almost totally eliminated ptau-217 in their plasma, tantalizingly suggesting that early treatment might prevent any further spread of aberrant tau aggregates in their brains. If that’s true, it’s a critical finding, because it’s aberrant tau that is most likely directly responsible for driving people into dementia: beta-amyloid is the gun, but aberrant tau is the bullet.
Lecanemab Too
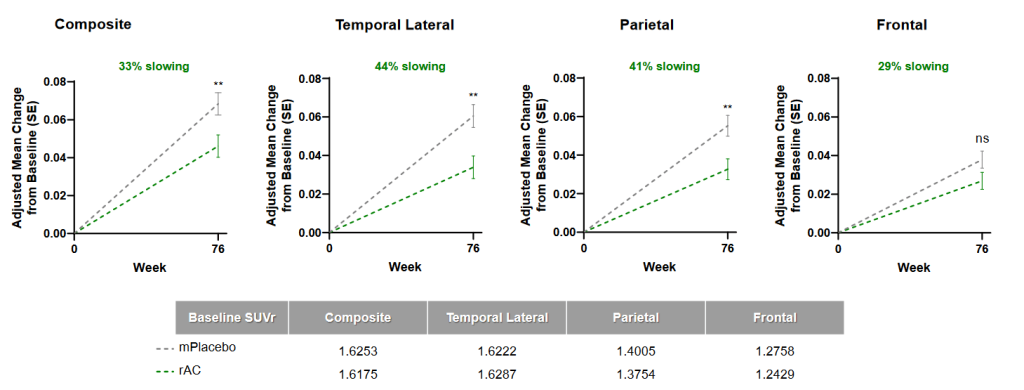
And remarkably, lecanemab not only slowed down cognitive decline more in people in the low-aberrant tau subgroup than in the overall substudy population, but appeared to actually claw back lost cognitive function! We can’t say this with complete confidence because the small numbers of people in the study created opportunity for artifacts. But it certainly looks as if people whose brains had relatively low levels of existing tau damage and who received lecanemab ended the trial with better cognitive function than they began with, as measured on the CDR-SB test.
That was a group average, so let’s break those results down. Over three in four of the people in the low-tau group who received lecanemab (76%) suffered no decline in CDR-SB, versus 55% of the low-tau aggregate group receiving placebo. And a remarkable 60% of the people in the low-damage lecanemab-treated group finished the trial with improved cognitive function compared to their baseline on this scale! (This compares to less than half as many (28%) of those with similar low burdens who got placebo infusions).
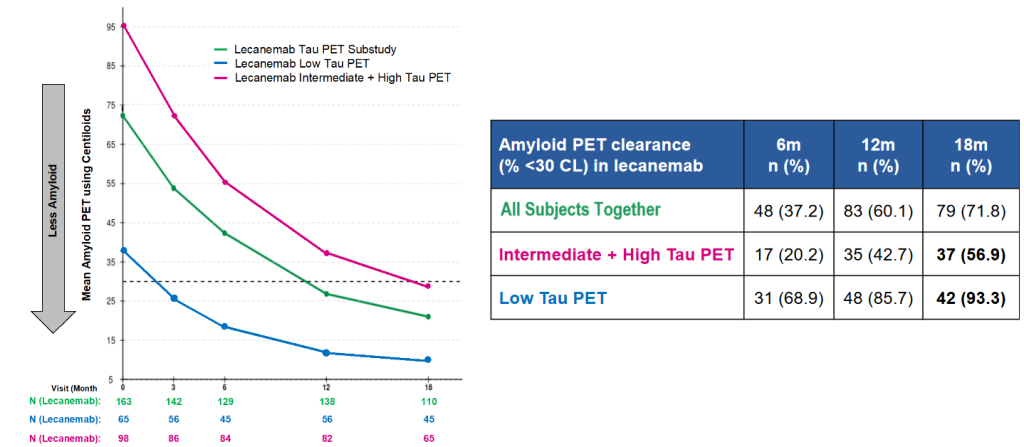
People with low aberrant tau burdens also appeared to have reversed their decline on a scale that combines cognitive function and the ability to carry out normal daily activities (ADCS-MCI-ADL) after they had been on the AmyloSENS therapy for six months. This may reflect a threshold level of clearance that it took lecanemab six months to achieve, or that it takes a while for those functions to recover after lecanemab clears amyloid and exerts its downstream effects on other damaging processes. Indeed, if you follow the trajectory of the graph, it is tempting to wonder if people with a low initial amassment of aberrant tau might have eventually gotten back to better life function than they had before they even started receiving treatment!
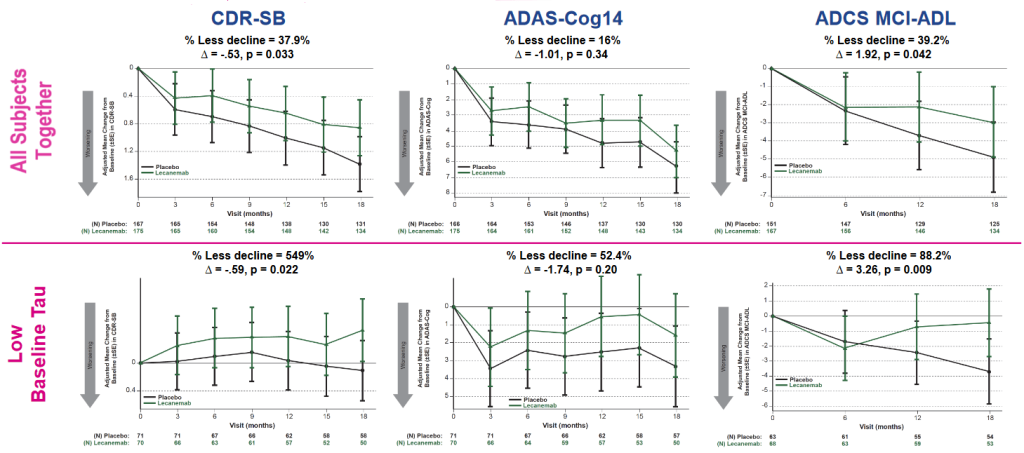
Lecanemab also slowed the downstream march of tau aggregates across the brain, with a stronger effect in the volunteers who started treatment with low levels of aberrant tau in their brains. Donanemab also had an impact on aberrant tau spread, but seemingly only in those in whom it cleared beta-amyloid the most quickly. If it’s correct that lecanemab was more broadly effective in applying the brakes on further tau spread, it might be because of the particular beta-amyloid assemblies that it targets. Lecanemab captures soluble oligomers and protofibrils, which seem to play a larger part in disrupting tau from its normal activities, whereas donanemab was engineered to go after a species enriched in insoluble amyloid plaque.
The Tortoise (on Steroids) Beat the Hare
Researchers at CTAD also presented evidence for the benefit of clearing beta-amyloid early on from the long-term extension of a trial called DIAN-TU. This trial started one step back from the Phase III trials for lecanemab and donanemab we’ve been discussing so far. As a reminder, everyone in those trials had already suffered at least the level of cognitive decline that is euphemistically referred to as “mild” cognitive impairment (MCI), and many were in the early stage of clinical dementia. By contrast, the DIAN-TU trial was aimed at prevention in a profoundly high-risk group of people: individuals with inherited mutations that virtually guaranteed that they would develop AD by their 50s and who already had beta-amyloid detectable in their brains by PET, but whose memory and planning ability still seemed to be intact.
The researchers initially randomized volunteers to receive either one of two beta-amyloid-targeting antibodies (gantenerumab or solanezumab) or placebo for an average of 5 years. At the scheduled end of the trial, gantenerumab seemed to have had no benefit — but the subjects were all given the opportunity to join an extension study in which they would all get gantenerumab while scientists followed up with them to track the long-term effects of treatment.
Gantenerumab has a chequered history. It failed an initial trial in people who were in the early stages of AD (similar to the subjects in the lecanemab and donanemab Phase III trials), but it was then resurrected — only to fail again in a similar trial that was published while this blog post was being written (though the company had announced its foundering months previously).
Why did gantenerumab fall short where two other AmyloSENS therapies (plus probably aducanumab/Aduhelm®) succeeded? Partially, it may have been because gantenerumab attacks full-length beta-amyloid fibrils, which are a late and more stable form of beta-amyloid aggregate, whereas (as we discussed along the way in a previous post) the best target for AmyloSENS therapies is probably soluble beta-amyloid oligomers and protofibrils (which lecanemab targets).
Another important factor was likely the low doses of gantenerumab used in the two (or three) Phase III trials. The original Phase III used 105 or 225 milligrams once every 4 weeks, and twinned ‘mulligan’ Phase IIIs only gradually built up to 510 milligrams. The researchers were being cautious about side-effects, but it’s clear in hindsight that even the 510 mg dose was too low. As the Phase III trial report explains, only about a quarter of the volunteers in the Phase III trial achieved the amyloid-negative status that was attained by everyone treated with donanemab in its successful Phase III trial and by over half of the people treated in the lecanemab trial.†
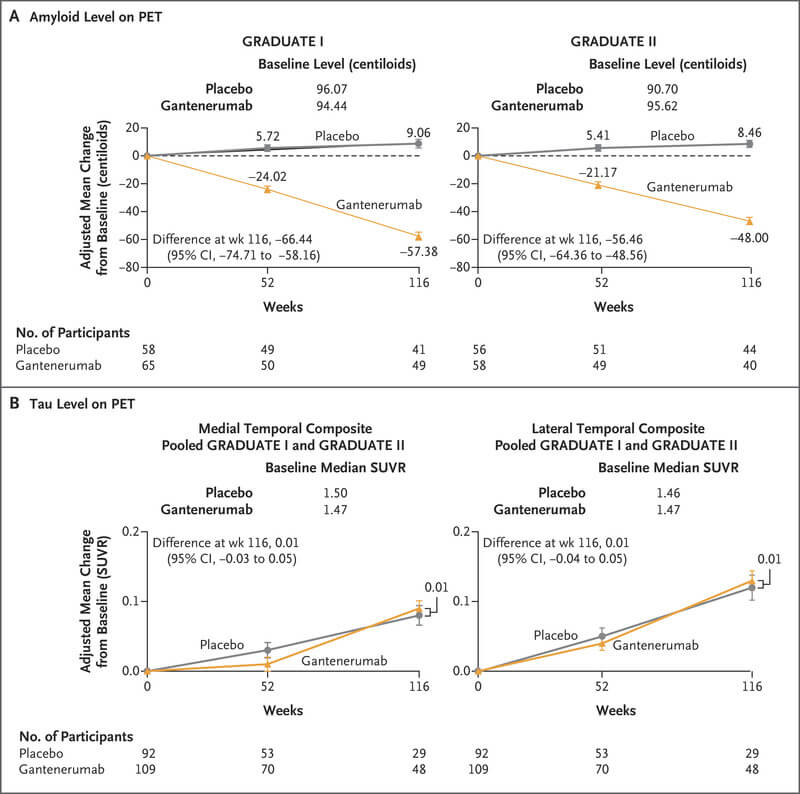
So while it would have been understandable if Roche had rounded up all its gantenerumab and consigned it to the biological waste barrels, it was still reasonable to think that gantenerumab might still work as effectively as the other antibodies if administered at high enough doses for long enough —especially if given earlier than in the Phase III trial.
So they took a chance, upped the dose, and the DIAN-TU prevention trial rolled on.
The long-term DIAN-TU prevention study that was presented at CTAD started volunteers off on more than twice the dose used in the rebooted Phase IIIs (1200 milligrams) — and when they signed these same volunteers into the extension study, they increased the dose yet again, bringing it up to two and a half times that dose (3000 milligrams)!
By this point, the extension study had become quite a tangled ball of biomedical yarn. For five years, one group had been treated with gantenerumab for an average of five years during the trial itself; a second group had instead received solanezumab; and a third group had received placebo infusions. And then after a one-year gap imposed by the COVID immunosenescence pandemic, all of those who signed up for the extension got gantenerumab for an additional two years, at two-and-a-half times the dose used in the original randomized phase of the trial. Thus, people who started the trial on placebo had received gantenerumab for just the last two years of the extension study, while at the other extreme, others had received it throughout the study as a whole, leading to an average of eight years of treatment (again, with a one-year hiatus).
The researchers compared the cognitive paths of all of these groups combined to the trajectories of placebo-infusion recipients who did not join the extension study — that is, people who effectively had been untreated from the beginning of the trial to the end of the extension study. When the researchers rolled all of the groups in the extension study together and compared them to these controls, it wasn’t clear whether the two years of gantenerumab treatment tacked on at the end had done anything to prevent the onset of AD symptoms based on CDR-SB.
This is an understandable outcome, since most of the people in the extension had been on something other than gantenerumab for the first six years or so. People originally on placebo would simply have suffered five or more years of the “normal” collision course with dementia — and even those who received five years of solanezumab might not have been any better-placed to take advantage of the final year of gantenerumab. Solanezumab failed in Phase III testing, and failed again in a very early-intervention AD prevention trial. While many things may have been involved in these flops, the reason most important to this newer trial is that solanezumab predominantly binds single monomers of beta-amyloid rather than beta-amyloid aggregates, which is at minimum an inefficient way to reduce the pathological aggregated species and at worst may even interfere with an antimicrobial role for beta-amyloid. And even when it captures these monomers, solanezumab may not even reduce the overall level of beta-amyloid monomers in the brain. Instead, evidence suggests that it recirculates them and then drops them right back off in the brain!
But a very different picture emerged when the researchers looked at each subgroup individually. People who were treated with gantenrumab from the very beginning and then into the extension were only half as likely to develop AD as measured by CDR-SB as people in the “all-placebo all the time” group! And even “all-gantenerumab” volunteers who did develop AD symptoms by the end of the extension enjoyed six extra years free of their genetically-fated dementia based on their estimated year of onset. And while the numbers were small enough that we can’t be confident in them, it appeared that even the volunteers who had originally been on placebo or solanezumab enjoyed a delay of about three years after their two extra years on gantenerumab, even though they a similar fraction of them wound up with AD in the end as in the all-placebo controls.
The Marathon and the Decathlon
It bears repeating something we’ve said before. To qualify for the Phase III trials for aducanumab, donanemab, lecanemab, and gantenerumab, participants needed to exhibit cognitive dysfunction severe enough to indicate that they had already lost about one-third of the neurons in the entorhinal cortex outright.‡ Their brains would likewise need to already be ravaged by the aging damage most responsible for neurodegenerative aging of the Alzheimer’s type (beta-amyloid and in some cases aberrant tau).
On top of that ruin, we know from other research that people suffering from AD also have more neurons hobbled by mitochondria bearing deletion mutations than other people of the same age and an elevated burden of senescent neurons in their brains.
These trials provide robust evidence supporting the classical “Amyloid Cascade” — the idea that neurodegenerative aging of the Alzheimer’s type is an agonizing domino-tumble of destruction that starts as beta-amyloid drives tau aggregates to invade additional regions of the brain, leading to neurons first failing to interact with each other and eventually dying, all culminating in dementia. Scientists originally built the cascade model by thoughtfully piecing together work done in cells in Petri dishes, transgenic animals, and the autopsied brains of people who had died with and without an AD diagnosis during their lifetimes. More recent research confirmed the cascade in ever greater detail thanks to new imaging tools that allow scientists to see beta-amyloid and other kinds of aging damage in the brains of living people who are succumbing to aging and the slide into AD.
And now the trials demonstrate what the cascade model always implied. When doctors use these new AmyloSENS therapies to clear beta-amyloid out of people’s brains, they are able to slow or stop the further accumulation of downstream damage and rig a drag chute on the brain’s careening crash-car course. Specifically, these new data from the CTAD conference show that the earlier in the cascade you intervene, the more of that further downstream damage you can prevent from accumulating — and the more you can slam the brakes on cognitive decline. And in the DIAN-TU study, it appeared that treating people after some beta-amyloid and tau aggregates had accumulated (but before impaired cognitive functioning set in) could even prevent dementia in some people who were otherwise doomed to early AD by mutations. Additionally, for those in whom AD set in anyway, they had a longer runway before crossing the threshold of AD.
Following this logic, it seems increasingly likely that clearing out beta-amyloid early enough might forestall AD for decades. That’s the marathon approach: intervene in outwardly healthy middle-aged people with seemingly intact brains, and keep treating them indefinitely to save their minds.
DIAN-TU is testing the marathon approach in people who are extremely susceptible to AD — to extend the metaphor, who are running their entire race uphill. But there are now two “marathon” trials in people whose risk of AD is driven by aging rather than defective genetics. One is the AHEAD Study, which will test lecanemab in volunteers 55 years of age or older who have either an intermediate level of beta-amyloid in their brains (in one trial) or a higher level (in the other), but whose cognitive function is typical for their age. The other trial is TRAILBLAZER-ALZ 3, which will test whether donanemab can prevent older volunteers (aged 65-80) who are cognitively intact but have serum markers of abnormal tau from sliding into dementia.

These trials are some of the very first opportunities for people who are not already suffering disabling age-related disease to get access to an effective rejuvenation biotechnology — or (if they wind up on placebo) to at least get cutting-edge molecular aging damage diagnostics and help accelerate the progress of damage-repair longevity therapeutics into wider use. People email SENS Research Foundation every month asking to volunteer for experimental longevity treatments: if you qualify, here is your chance.
That’s the marathon. For those of us who are already past this threshold — or who will be by the time that doctors start routinely using these AmyloSENS therapies in this aggressive way — there is the decathlon. Yes, purge the brain of beta-amyloid — but when an aging brain has already endured much of its downstream effects, go after those targets too. In addition to slowing the further spread of aberrant tau inside neurons by removing upstream amyloid, we need rejuvenation biotechnologies to degrade it inside the neurons themselves, as SENS Research Foundation scientists are working to do. (This is different from what nearly all of the many tau immunotherapies that drug companies are currently developing do, which is to capture these aggregates as they spread outside the neurons. This approach is like only killing the cockroaches you spot scurrying across your kitchen floor instead of hunting down and poisoning their nests. We have recently seen the latest of several failures with such extracellular antibodies).
And since people with even “mild” cognitive impairment have already lost a large fraction of the neurons of their entorhinal cortex by the time of diagnosis, we will need to replace those neurons with new ones and reinforce the remaining neuronal circuitry to sustain its function against loss to tau aggregates or any of the other slings and arrows of aging. Dr. Jean Hébert has developed an ingenious neuronal replacement strategy that (so far, in mice) can fan replacement cells out across the brain. The plan is to turn these engineered cells into neurons on-site; SRF has been funding this bold and critical project from the time Dr. Hébert first proposed it until today.
There is also other, less-AD-specific cellular and molecular aging damage in the aging brain that afflicts people with AD even more than people of the same age without that diagnosis. Removing, repairing, replacing, or rendering harmless this more broadly age-related will be essential to preserving the brain regardless, and can be expected to contribute to holding off AD.
For instance, the brains of people with AD are riddled with senescent cells, and destroying senescent cells in the brain has been shown to extend cognitive function and reduce other damage in the brain in animal models of Alzheimer’s disease. Now the senolytic combination dastatinib plus quercetin (D+Q) is being tested in human AD patients. In a very small, uncontrolled 12-week trial, dasatinib (but apparently not quercetin) got into the fluid bathing the brain and spinal cord (CSF), which suggests that “D” at least would reach the brain. The senolytic treatment lowered levels of several SASP factors in the CSF, as well as increasing levels of a particular species of beta-amyloid.
That may sound bad, but levels of this molecule predictably decline in the CSF as a person gets closer to AD dementia (at which point the level plateaus). This is perhaps because these molecules increasingly disappear from the CSF as they stick together and lodge in the brain. So a rise in this molecule may actually indicate a movement back to normal mobilization of amyloid out of the brain and out through the bile. This species of beta-amyloid also rises in the CSF of people treated with amyloid-clearing antibodies, and in that situation the rise is unlikely to be explained by the beta-amyloid that is actually in the vise-grip of the antibodies themselves.
Curiously, the senolytic treatment also increased CSF levels of the signaling molecule interleukin-6 (IL-6), which is a classic SASP factor: the investigators speculate that this may be released by dying senescent cells, or may be released to summon immune cells to clear out the debris of senescent cells destroyed by the drugs.
Another kind of aging damage that AD patients suffer disproportionate to their age is neurons overtaken by mitochondria bearing deletion mutations in their brains. Rendering mitochondrial mutations harmless is likely to benefit our brains along with all other organs regardless, but this AD-specific excess comes on top of the background effects of aging in all of us cry out for a biotechnological solution. This is a much more challenging problem than the AmyloSENS therapies that are the focus of this post, but SRF scientists are working diligently to crack it.
We also know that other kinds of aging damage and their downstream consequences increase a person’s risk of dementia overall: you only have one brain, and clinical dementia happens because of the sum of all the different outrages that aging throws at it, whether it’s diagnosed as AD or something else. So repairing that seemingly unrelated damage — even damage to structures outside of the brain itself — will also help us maintain our ability to remember, think, and plan sharply.
For instance, one of the accepted causal risk factors for dementia is hypertension, which contributes to dementia through the risk of strokes (including transient ischemic attacks and “silent strokes”) and damage to the small blood vessels and the insulating material for the “wiring” that links our neurons together into functional circuitry. Much of the rise in systolic blood pressure with age is driven by damage to the long-lived structural proteins in the artery. Breaking excessive and abnormal crosslinking in the aging artery and repairing damage to the elastin fibers could allow our arteries to expand youthfully when blood surges down the vessels after a heartbeat, greatly reducing the downstream damage. Breaking inappropriate crosslinks might also counteract much of the dementia risk driven by diabetes.
Atherosclerosis is also a critical factor in most strokes, and we can attack it directly by degrading or removing damaged cholesterol from the foam cells in our arteries. Two longevity biotech startups are each pursuing distinct strategies on this front as we speak: Cyclarity Therapeutics is soon to enter human clinical trials with its novel cyclodextrin dimer UDP-003, which their data suggests can smuggle damaged cholesterol products out of foam cells. Meanwhile, Repair Biotechnology has shown in animal models that its Cholesterol Degrading Platform can degrade damaged or simply excessive cholesterol inside the foam cell, dramatically reversing atherosclerosis in animal models.
Age-related hearing loss has also been linked to dementia risk, possibly indirectly by reducing mental and social stimulation when people can’t follow a conversation, or even directly via loss of neural stimulation. There are caveats to these findings in a recent study of the brains of people with hearing loss and a clinical trial that assigned people randomly to receive hearing aids or health education, but replacing hair cells lost to age could keep us keen to our friends’ laughter and engaged with the issues of the day, thereby helping preserve and extend our cognitive function.
Ultimately, these therapies will work in concert to delay, slow, arrest, and eventually reverse AD and other forms of neurodegenerative aging. As each rolls out, we can use it to buy time, extending our runway while researchers build more components, until eventually our cognitive Space Shuttle can achieve escape velocity and rocket into the stars.
Cognitive Escape Velocity
Some readers may be thinking that from what I’ve said above, there’s no hope for them or their loved ones unless either they start receiving one of the successful AmyloSENS therapies immediately or we develop a long laundry list of (cognitive) rejuvenation biotechnologies within a few short years. Certainly earlier or more comprehensive damage repair is better than their opposites, and one way to address this concern is to consider enrolling in the AHEAD Study or TRAILBLAZER-ALZ 3.
But even for those who don’t, far more people’s minds can still be saved than would be suggested by that inference, because of the same principles behind longevity escape velocity. Whereas conventional risk-factor management drugs and “messing with metabolism” geroscience drugs can only slow down the rate at which aging damage is inflicted, SENS “damage-repair” longevity biotherapeutics actually remove existing damage from the brain or other tissues. This allows you to literally make up for lost time by bringing your brain’s burden of aging damage back to that of a younger person. This feature also buys time for additional (cognitive) longevity therapeutics to be developed that are either more effective than earlier iterations, or that remove, repair, or replace aging lesions for which there were no effective therapies when you first stepped on the launchpad.
These AmyloSENS therapies alone don’t provide a definitive solution to keeping our brains young — but they give an updraft to our cognitive glider, keeping us aloft for longer until we can finally fly under our own power.
* I don’t know why they initially chose to compare those with moderate levels with the overall population (moderate plus high) instead of the more granular analysis presented at CTAD; I speculate that it may have been because there were so few subjects with very low and very high levels that they worried it would wash out in statistical testing.