

SENSible Question: Some scientists have reported that viruses, bacteria, and other pathogens may help drive Alzheimer’s and other neurodegenerative diseases, and that the body uses beta-amyloid protein to fight them off. So doesn’t that mean it’s a bad idea to remove Abeta from the brain?
This is an important question, not only because of what it tells us about the specific issue at hand, but because it gets to the heart of the SENS “damage-repair” approach to longevity therapeutics.
Abeta is of course the sticky protein whose aggregates accumulate in the aging brain — most prominently in the brains of people diagnosed with Alzheimer’s disease (AD), and less dramatically in other dementias of aging. In fact, even absence of specific “diseases” of aging, Abeta is strongly implicated in driving so-called “normal” age-related cognitive decline. Accordingly, Abeta is an important target for therapies that remove damaged proteins from the brain, and part of the suite of rejuvenation biotechnologies that will be needed to keep our minds sharp, clear, and curious throughout greatly-extended life expectancies.
But as the questioner says, there is also mounting and important evidence that infectious microbes may be one of the drivers of Alzheimer’s and other diagnosed neurodegenerative diseases of aging. There’s lots of “scene of the crime”-type evidence for this: most people over the age of 65 have viruses and other microbes in their brains, and the brains of people diagnosed with Alzheimer’s disease have disproportionately high burdens of bugs like oral Herpes virus (HSV), the gingivitis bacterium P. gingivitis, and even the microbes that drive Lyme disease and syphilis. HSV in particular is disproportionately found in areas of the brain with high burdens of Abeta plaque.
And in the last 5-10 years, scientists have begun reporting that these two seemingly-unrelated drivers of neurodegenerative brain aging — Abeta aggregates and infectious pathogens — are actually interconnected in a surprising way. Evidence has been mounting that Abeta may actually be an antimicrobial peptide (AMP) — one of a class of small proteins that are produced by humans and other living things, and that exert broad, potent activity against bacteria, viruses, and fungi.
Dr. Rudy Tanzi, who discovered the genes behind several kinds of genetically-driven neurological disorders, reported in 2016 that Abeta binds to structures on the surface of eight microbes that infect humans. Binding to these sites on microbial invaders triggers individual molecules of Abeta to stick together, forming aggregated structures that imprison the pathogens.
To see if this effect was enough to actually protect animals from infection, Tanzi’s team inserted genes into several different species of lab animals that caused them to Abeta (which most animals don’t do naturally). Sure enough, the ability to produce Abeta significantly protected the animals from different infectious pathogens.
Then, when Dr. Tanzi and colleagues intentionally infected the brains of a mouse model of AD with a species of Salmonella bacteria, Abeta molecules rapidly locked into place together, with the highest levels of aggregated Abeta deposits at the site of infection. They also showed that Abeta had similar activity in three-dimensional cultures of infected human neurons.

These and related findings by Dr. Tanzi and others led to a new model for the role of Abeta in the aging and AD brain. In this new model, brain neurons produce Abeta as a way to protect themselves from microbial assailants. When they come in contact with a pathogen, molecules of Abeta bind to the intruder, which triggers them to stick together into aggregates. Trapping the brain bugs in a sticky web allows Abeta to deactivate the microbial raiders, protecting the brain from infectious assault.
Recognizing the Jigsaw Picture
With this new model, a number of things that scientists have been reporting for years suddenly start to make sense. For one thing, it’s long been known that the complement system is activated in the early stages of Alzheimer’s disease. The complement system is a part of the innate immune system that directly destroys pathogens by tearing open their membranes, and it was already known to be activated by other AMPs. But scientists have repeatedly reported that Abeta can also activate the complement system, which seemed like a bizarre, perverse thing for it to do. But that’s exactly what Abeta would be expected to do if it were itself an AMP: unleash dormant complement proteins at the location where they have trapped the intruding microorganism to help them finish the enemy off.
The new model also explains why proteins that are part of the complement system are often found bound up with Abeta plaques in the brain. Since the plaque handcuffs the very microbes that set off Abeta aggregation in the first place, he complement proteins wind up at the same location because the microbes they’re attacking are located there.
If Abeta is an AMP, it also reframes the role of inflammation in the aging and AD brain, and the associated activation of brain-resident immune cells called microglia. Microglia are like the macrophages of the brain, gobbling up particulate matter, cellular debris, and other harmful materials in the brain — including, importantly, Abeta — and digesting it in their lysosomes. If we reframe Abeta as an AMP, then the above progression may be somewhat analogous to the interaction of senescent cells with the immune system. Microglia actually have receptors on their surfaces that cause them to spring into action when they get a whiff of activated complement proteins, and as we mentioned above, Abeta causes dormant complement protein precursors to be converted into their active forms. In the Abeta-as-AMP model, this becomes an elegant host defense system: Abeta is released, traps a marauding microbe in a self-aggregating web of proteins, and then activates complement to help finish off the enemy and to recruit microglia to clean up the battlefield.
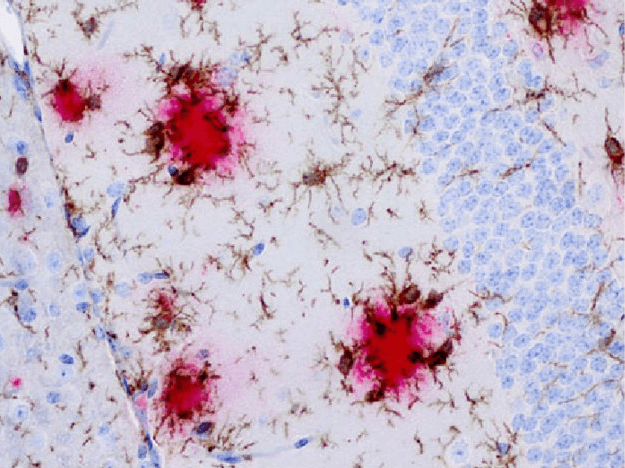
This sequence protects the brain from these toxic materials in the short term — first from the infectious intruder, and then from Abeta itself. But after years of taking in more and more Abeta and other harmful material in the aging brain, microglia eventually undergo a sort of Jekyll-to-Hyde transition. No longer able to take on more damaging material, the microglia nonetheless continue to senselessly churn out inflammatory factors, and may even turn traitor, attacking the very brain that they exist to defend.
If You Want War, Prepare for Peace
So your question is: if Abeta is helping to protect the brain from the threat of infection, doesn’t that make it potentially counterproductive to remove Abeta from the brain? Wouldn’t you be effectively scuttling a key brain defense system, leaving the brain even more vulnerable to infectious assault and neurodegeneration?
Indeed we would be — if our strategy were to inhibit the production of Abeta. Indeed, with benefit of hindsight, that might explain the fate of the many failed drugs that were intended to treat or prevent AD in exactly this way.
A few such drugs failed in clinical trials for unrelated reasons — notably, some of them were toxic to the liver, skin, or other organs. And Flurizan, which got further along than many of the drugs of this type, flopped because it failed to improve AD patients’ mental functioning, quality of life, or ability to take the most basic care of themselves.
But that’s the least of it. Most of the Abeta synthesis inhibitor drugs failed in clinical trials not because they merely didn’t work, but because they actually made cognitive function even worse in people with Alzheimer’s! This was true of verubecestat, lanabecestat, atabecestat, and most famously Eli Lilly’s semagacestat. In light of the new research about Abeta’s role in protecting the brain from microbial marauders, we are confronted with the possibility that part of the reason that these inhibitor drugs impaired cognitive function in people with AD might be that they were unwittingly disabling one of the brain’s critical self-protection systems.
Break the Eggs to Make the Omelet. But Clean Up the Pan.
But remember, inhibiting a metabolic function like the production of Abeta is exactly the kind of thing that SENS rejuvenation biotechnology never does! This is the key feature that sets the “damage-repair” approach to developing longevity therapeutics apart from the earlier “gerontological” (or, often now, “geroscience”) approach, as well as from the ultimately futile “risk factor management” approach that dominates current medical efforts to hold diseases of aging at bay.
Drugs that inhibit Abeta production are in fact classic examples of the “gerontological” approach. By stimulating or suppressing the metabolic processes that contribute to aging damage, “gerontological” therapies aim to dampen down the amount of damage that they produce (or, alternatively, ramp up processes that rectify the early, reversible steps leading from such processes to stable structural damage), and thus slow the accumulation of the damage that metabolic processes cause. By contrast, SENS rejuvenation biotechnologies are targeted directly at the damage itself, leaving the metabolic processes that produce it to proceed without interference — but cleaning up the ensuing damage before it accumulates to high enough levels to cause us harm.

One of the main reasons to favor the SENS approach is exactly because interfering with metabolic processes is risky, since all such processes are in one way or another essential to keeping us alive and healthy. There is no “aging program:” evolution did not build Abeta or LDL particles or the senescence machinery into us as so many little time bombs designed to ensure that we suffer degenerative aging. Instead, aging happens because the processes that were evolved to keep us alive and healthy in the short term are messy, and leave behind cellular and molecular damage in our tissues that our bodies don’t fully repair. In the long term, that damage accumulates in our tissues to the point that it impairs the ability of those tissues to carry out their exquisitely-regulated, evolved function. The ensuing dysfunction of a specific tissue is exactly what doctors then diagnose as a particular “disease of aging.”
In this case, Abeta is produced in the short term as an emergency response to microbial marauders; microglia are then activated and recruited to clear the dead pathogens and aggregated proteins out of the brain so that they don’t cause us harm of a different sort. So long as this cycle is executed flawlessly, the brain remains protected from threats and sustains function. But none of these processes less than perfect, they leave behind a few microbes here … a few protein aggregates there … and a few dysregulated microglia in another corner.
Meanwhile, other aging processes make it increasingly difficult to close the loop on the cycle of releasing and aggregating Abeta, destroying pathogens, and recruiting microglia to clean up the battlefield afterward. For instance, damage to the protective barrier shielding the brain and an increasingly dysfunctional immune system with age allow increasing numbers of microbes to penetrate the brain. And as microglia take up more and more Abeta over time, their lysosomes become increasingly unable to process the waste being sent their way, similar to what happens in arterial macrophages in the development of atherosclerotic cardiovascular disease. As a result, more and more Abeta aggregates get left behind to increase brain inflammation and weaken the connections between brain cells, and microglia become increasingly dysfunctional, ultimately doing more harm in the brain than good.
The core of this is articulated by Dr. Tanzi himself:
Abeta first entraps and neutralizes invading pathogens in beta-amyloid. Abeta fibrillization drives neuroinflammatory pathways that help fight the infection and clear beta-amyloid/pathogen deposits. In AD, chronic activation of this pathway leads to sustained inflammation and neurodegeneration. …
In the antimicrobial protection model, the modality of Abeta’s pathophysiology is shifted from abnormal stochastic behavior toward dysregulated innate immune response. However, beta-amyloid deposition in AD still leads to neurodegeneration. Thus, the new model extends but remains broadly consistent with the Amyloid Cascade Hypothesis and overwhelming data showing the primacy of Abeta in AD pathology.
As usual, the SENS approach is to cut the Gordian knot by taking a “hands-off” approach to the regulation of Abeta production, complement proteins, and the other metabolic processes implicated in the brain’s attempts (sometimes misguided) to defend itself, and instead to directly remove Abeta aggregates after they’ve been formed.
Remember, Tanzi discovered that Abeta defends the brain against microbial invaders by forming aggregates that capture and neutralize them. Once they’ve already carried out the attack, the whole snarled-up mess — Abeta polymers, dead microbes, and complement proteins— serves no further purpose and can be toxic to the brain. So Abeta that is cleared out after becoming aggregated has already finished serving a useful purpose, and is mere battlefield rubble that must be safely swept away to help rebuild the neighborhood.
All the Damage, All the Time
A comprehensive damage-repair strategy to sustain brain function over time will involve more than just clearing out Abeta, of course. SENS Research Foundation scientists are working right now to develop rejuvenation biotechnologies that directly degrade aberrant tau inside of neurons, and we are funding work to replace lost brain neurons and reinforce neuronal circuits.
And in the coming months, we expect to be able to announce a new project that promises to tackle the apparent therapeutic dilemma posed by the Abeta-as-AMP model even more pre-emptively: by directly destroying viruses and bacteria in the brain, while holding Abeta in a safe “reserve” mode. This is again a case of attacking the actual damage in the aging body (in this case, brain microbes), which can be contrasted with the recent failure of Cortexyme’s Atuzaginstat and abandonment of the next-generation COR588 —drugs that were intended to block gingivitis bacteria from contributing AD by breaking up their toxic metabolites, but that didn’t address the pathogen that was releasing those toxins in the first place.
A full strategy for indefinite brain maintenance may also one day involve fortifying the lysosomes of microglia to greatly increase their ability to safely clear out Abeta and prevent them from becoming dysfunctional and “flipping” on the brain, attacking the neurons they exist to protect.
What we’re not going to do is interfere with the brain’s ability to produce Abeta in the first place, or prevent it from aggregating as some have proposed. This has been true from the beginning, and now we see in hindsight another reason why such “messing with metabolism” is ill-advised: these approaches would risk leaving microbes free to vandalize the aging brain unchecked.
It's Not a Bug — It’s a Feature
Similar kinds of therapeutic pitfalls ensnare scientific proposals to interfere with metabolic processing driving other kinds of aging damage. Take senescent cells. It’s very clear that the accumulation of senescent cells in our tissues over time drives aging and age-related disease. This has frequently tempted scientists to develop drugs that might prevent cells from becoming senescent. But cells don’t pull on their senescence-inducing fire alarms as a kind of perverse metabolic prank. The elaborate machinery that turns cells exists to perform essential functions as part of embryonic development, wound healing, and the preventing fibrosis after injury, and most importantly in shutting down replication of cells on the verge of becoming cancer. The problem is not cells becoming senescent in the first place, it’s that a subset of them persist and accumulate after that essential function has been completed.

The solution to this problem is not to prevent cells from undergoing senescence: that would leave us vulnerable to unchecked cancer and fibrosis. Instead, the SENS approach is allow cells to undergo senescence when they need to — and then destroy senescent cells before they can accumulate to the point of doing us irrevocable harm.
Similarly, our cellular power plants (mitochondria) generate toxic free radicals due to the imperfection of the machinery that converts energy derived from food to cellular energy in the form of ATP. This causes the mitochondria to damage themselves, including causing large deletions in their DNA, which forces the cell into an alternative energy regime that promotes damage and aberrant signaling in surrounding cells. Some of the “gerontological” mindset have proposed using drugs to dissipate the gradient that keeps the cellular energy production machinery working at full capacity as a way to keep mitochondria from damaging themselves so much and perhaps hold off mutation for a little longer. SRF scientists are instead working on to engineer backup copies of the mitochondrial genes, so that our mitochondria can continue to produce energy normally, even if they do inflict mutations on themselves.
And so on, whether it’s aggregates inside cells, or nuclear DNA mutations and stable epigenetic damage, or the loss of functional stem cells. The “damage-repair” approach allows metabolism to do its essential work, but severs the link between metabolism and pathology by cleaning up the damage afterward. This iterable approach will allow us to live free of age-related disease and debility — potentially indefinitely — without compromising what not only keeps us alive, but makes life worth living.