
As noted in a previous post, the clearance of neurofibrillary tangles (NFT) and other intracellular aggregates is a key rejuvenation biotechnology to restore youthful function to aging brains, especially those with Alzheimer’s disease (AD) and a range of other age-related neurodegenerative disorders. While Phase I and II clinical trials with the original, active beta-amyloid (Aβ) vaccine AN1792 have demonstrated a wide range of benefits in patients exhibiting a sufficiently robust immunological response, autopsy studies in such patients have reported no evident effect on mature tau neuropathology (NFT and neuropil threads); this seems likely to be a significant barrier to the the potential benefits of Aβ clearance, consistent with the limited cognitive benefits observed in these trials.
However, as we discussed in an earlier post, a recent study(1) did find, amongst other things, a substantial restoration of normal curvature in immunized patients’ neuronal processes, accompanied by significantly fewer hippocampal neurons stained with paired helical fragment-1 (PHF1) antibodies, which target a range of hyperphosphorylated tau (phospho-tau) species. These effects were not, however, accompanied by parallel reductions in staining by Alz50 antibodies (recognizing tau phosphorylated at serines 199 and 202 and threonine 205) or Thioflavin-S, suggesting that AN1792 can remove early-stage, but not more mature phospho-tau aggregates.
While suggestive, this study(1) was limited by a small patient base (n = 5) and minimal therapeutic exposure: the Phase II trial in which they had participated was halted due to side-effects (meningoencephalopathy in ~6% of immunized patients) after patients had received only two doses of AN1792. Now, a new study has more strongly reinforced and expanded this finding.
The new report(2) compared neuropathology at autopsy in 10 AD subjects (iAD) immunized with AN1792 in the earlier Phase I trial, to 28 untreated control AD (cAD) subjects. Unlike the subjects in the earlier report (1), these subjects had received 5-7 sessions of vaccine over the course of treatment.
The phospho-tau load was lower in the iAD than the cAD group in the cerebral cortex (cAD 1.08% vs. iAD 0.72%, P = 0.048), CA1 hippocampus (cAD 2.26% vs. iAD 1.05%; P = 0.001), subiculum (cAD 1.60% vs. iAD 0.31%; P = 0.001) and entorhinal cortex (cAD 1.10% vs. iAD 0.18%; P < 0.001). … Aβ immunotherapy-associated reduction was confined to neuronal processes, i.e. neuropil threads and dystrophic neurites. … There was a significant correlation between phospho-tau dystrophic neurite cluster counts and Aβ42 load in the cortex (r = 0.879, P = 0.001) but not with phospho-tau load or phospho-tau-positive neurons. This correlation is consistent with the localisation of dystrophic neurite clusters to plaques. … However, the phospho-tau accumulation in the neuronal cell bodies, contributing to neurofibrillary tangles, appeared not to be affected. …
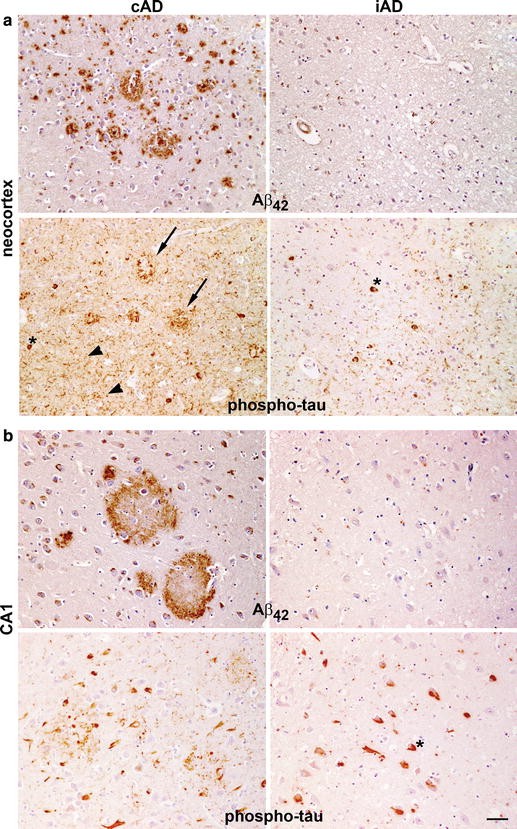
Figure 1. Examples of “the cerebral neocortex (a) and CA1 hippocampus (b) of Aβ42 (21F12 antibody) versus phospho-tau (AT8 antibody) staining of an unimmunised AD control (cAD) and an immunised AD case (iAD). From (2).In probing the mechanism of clearance of early tau pathology in dystrophic neurites, the authors note the possibility that this could be due to the actual phagocytic removal of the phospho-tau-laden dystrophic neurites themselves. Their findings suggest otherwise, however, as
we have not observed any evidence of phospho-tau within microglia, whereas Aβ is readily detectable within microglia in many of these immunised AD cases. This observation may be interpreted as suggesting that the phospho-tau is not cleared by phagocytosis. … An alternative explanation is that the neurons are able to mobilise the aggregated tau in the dystrophic neurites and remodel the neurites. The mechanism of clearance of phospho-tau is difficult to address directly in a study of this type. … However, experimental models with aggregation of both Aβ and tau do exist. In triple transgenic (3 × Tg-AD) mice early, but not the late, forms of phosphorylated human tau were removed by a single injection of anti-Aβ antibody into the brain [3]; reaccumulation of Aβ preceded reappearance of neuronal tau. In another model, active Aβ immunisation of APPSw/NOS2−/− mice, which develop Aβ deposits as well as hyperphosphorylated mouse tau, produced a significant reduction of deposits of both Aβ and hyperphosphorylated tau[4]. Our findings in human AD following Aβ immunisation are consistent with these experimental observations linking Aβ and tau pathobiology. … This is consistent with the amyloid cascade hypothesis which states that tau pathology is linked to and downstream from Aβ accumulation in AD and confirms the position of Aβ as a target for modifying both Aβ and tau aggregation in AD. (2)Indeed, recent studies(notably (5-7)) would seem to highlight an even more optimistic reading of the specific clearance of phospho-tau from neuronal processes. In a variety of systems, these reports suggest that Aβ-associated neurotoxicity and synaptic dysfunction may in large part be mediated by aberrant redirection of tau from its normal axonal localization to the somatodendritic compartment, even in the absence of any change in the total levels of tau or of axonal transport proteins. Importantly, lowering tau protein levels affords significant protection against such toxic effects, despite a lack of effect on baseline axonal transport. This is consistent with many previous studies (eg, (8-10)) showing that depletion of native or transgenic tau protects rodent models of AD against Aβ neurotoxicity, even absent an effect on NFT. These findings therefore suggest that the ability of Aβ immunotherapy to clear early tau pathology from the dentrites, as found in the current report,(2) may have an unexpectedly powerful neuroprotective effect, if initiated at a relatively early stage in the “amyloid cascade,” even if not accompanied by concomitant NFT clearance. Additionally, it seems reasonable to expect that the accumulation of NFT may be substantially prevented by such early immunotherapeutic clearance of Aβ, accompanied by clearance of its precursors in the dendrites. The new report does however seem consistent with a much more limited benefit of Aβ immunotherapy alone once the disease is well-esatablished, as in these patients and those in earlier autopsy reports:
One possible explanation for the continued decline in function [in AD patients after Aβ immunisation] may be the lack of protective effect of Aβ immunisation on accumulation of phospho-tau in the neuronal cell bodies, implying that additional therapy may need to be directed at neurofibrillary tangles. … Each of the iAD patients had cognitive function scores within the range MMSE score 15–25/30 at the time of their immunisation. This MMSE range is associated with Braak stages III–V whereas Braak stage VI is associated with MMSE scores in the range 0–10. The decline in MMSE score to 0 in all but one of the iAD cases prior to their death is therefore in keeping with progression in their Braak stage following Aβ immunisation. Correspondingly, the lack of difference in phospho-tau-positive neuron counts between the iAD and cAD cases suggests that phospho-tau continued to accumulate in neuronal cell bodies to reach, by the time they died, a level indistinguishable from that in unimmunised terminal AD. We cannot exclude an upstream role of smaller tau aggregates (not detected by AT8 staining) in the cognitive deterioration as observed in animal studies
In showing that Aβ immunisation can influence phospho-tau pathology, we confirm the position of Aβ as a target for modifying tau accumulation in AD and demonstrate a link between these proteins. However, the continuing progression of cognitive decline in AD patients after Aβ immunisation may be explained by its lack of apparent effect on tangles.(2)As the authors note, and as animal studies reviewed in an earlier post suggest, immunotherapy directed at clearance of tau pathology would seem to offer a way to overcome this limitation.
References
1. Serrano-Pozo A, William CM, Ferrer I, Uro-Coste E, Delisle MB, Maurage CA, Hock C, Nitsch RM, Masliah E, Growdon JH, Frosch MP, Hyman BT. Beneficial effect of human anti-amyloid-beta active immunization on neurite morphology and tau pathology. Brain. 2010 May;133(Pt 5):1312-27. Epub 2010 Mar 31. PubMed PMID: 20360050; PubMed Central PMCID: PMC2859150.
2. Boche D, Donald J, Love S, Harris S, Neal JW, Holmes C, Nicoll JA. Reduction of aggregated Tau in neuronal processes but not in the cell bodies after Abeta42 immunisation in Alzheimer’s disease. Acta Neuropathol. 2010 Jul;120(1):13-20. Epub 2010 Jun 9. PubMed PMID: 20532897.
3. Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM. Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron. 2004 Aug 5;43(3):321-32. PubMed PMID: 15294141.
4. Wilcock DM, Gharkholonarehe N, Van Nostrand WE, Davis J, Vitek MP, Colton CA. Amyloid reduction by amyloid-beta vaccination also reduces mouse tau pathology and protects from neuron loss in two mouse models of Alzheimer’s disease. J Neurosci. 2009 Jun 24;29(25):7957-65. Erratum in: J Neurosci. 2010 Jan 20;30(3):1197-8. PubMed PMID: 19553436; PubMed Central PMCID: PMC2871319.
5. Vossel KA, Zhang K, Brodbeck J, Daub AC, Sharma P, Finkbeiner S, Cui B, Mucke L. Tau reduction prevents Abeta-induced defects in axonal transport. Science. 2010 Oct 8;330(6001):198. Epub 2010 Sep 9. PubMed PMID: 20829454.
6. Zempel H, Thies E, Mandelkow E, Mandelkow EM. Abeta oligomers cause localized Ca(2+) elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. J Neurosci. 2010 Sep 8;30(36):11938-50. PubMed PMID: 20826658.
7. Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wölfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Götz J. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell. 2010 Aug 6;142(3):387-97. Epub 2010 Jul 22. PubMed PMID: 20655099.
8. Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science. 2007 May 4;316(5825):750-4. PubMed PMID: 17478722.
9. Oddo S, Vasilevko V, Caccamo A, Kitazawa M, Cribbs DH, LaFerla FM. Reduction of soluble Abeta and tau, but not soluble Abeta alone, ameliorates cognitive decline in transgenic mice with plaques and tangles. J Biol Chem. 2006 Dec 22;281(51):39413-23. Epub 2006 Oct 20. PubMed PMID: 17056594.
10. Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005 Jul 15;309(5733):476-81. PubMed PMID: 16020737; PubMed Central PMCID: PMC1574647.