

Short summary: A supporter asks if the Hallmarks of Aging could effectively be substituted for the seven categories of cellular and molecular damage in the SENS platform. The answer is ‘no.’ Part One of this two-part post discussed some of the broad conceptual reasons why not; in Part Two, we will compare and contrast the various Hallmarks and Strands of the SENS Seven one by one to see where the Hallmarks fall short.
Looking Through the Mugshots
In Part One of this two-part post, we looked at the thinking behind the structural framework into which the Hallmarks are organized. In this post, we will explore the Hallmarks individually and see how well they up with the SENS Seven — and when they don’t line up, or when they overlap with more than one of each other, we’ll ask why the two systems differ, and what might be missing or superfluous in either system.

Genomic Instability vs. OncoSENS
Both of these categories are centered on what the Hallmarks paper describes as “the accumulation of genetic damage throughout life,” but the subtle dividing line we’ve seen separating the two systems manifests here too. The Hallmarks paper frames the problem in terms of process: “genomic instability” refers to a progressive increase in genetic abnormalities in cancer and aging, and “genetic damage” can include reversible DNA lesions that may or may not mature into irreversible mutations. (Compare the references to “primary causes of cellular damage” and elsewhere “the physiological sources of aging-causing damage”). By contrast, the SENS strategy relentlessly focuses on bottom-line damage to the genome that the cell can’t fix on its own. In the case of OncoSENS, that means mutations (and epimutations — more on this later), not just transient DNA lesions.
If you frame the problem as “genomic instability,” it’s not clear what you can do about it. Every time a cell replicates its genes to divide and form a new cell; or opens up its DNA to create a “working copy” of a gene so it can make the protein that the gene encodes; or engages in one of the many biochemical processes that generate free radicals or other reactive byproducts and intermediates — in all such cases, there’s an opportunity for genetic damage.
The cell has DNA repair machinery that can fix some of these lesions, so you might think that you could solve this problem by developing drugs that stimulate that machinery. But our DNA repair machinery is energetically costly to the cell; it can come with surprising side-effects; it is mechanistically unable to fix certain kinds of DNA damage; and it can’t overcome the species-specific limits to the fidelity of the machinery. And perversely, the DNA repair machinery occasionally inflicts or makes permanent errors in the genetic code it’s trying to fix! In fact, errors made by the DNA repair machinery are one of the largest sources of mutations. And by definition, actual mutations are irreparable.
Since DNA maintenance is imperfect and comes with tradeoffs, and since we are nowhere near being able to repair existing genetic damage in the sense that SENS means by “damage” (i.e., mutations), Dr. de Grey’s insight was that for the critical leap forward into longevity escape velocity, it would be enough to render the effects of mutations and some other forms of genetic damage harmless to us.
The Hallmarks paper notes the three essential such effects in its discussion of the “genomic instability” Hallmark: cellular senescence, apoptosis (programmed “cellular suicide”), and cancer. SENS proposes to deal with these three by (respectively) terminating senescent cells (ApoptoSENS); using cell therapy (RepleniSENS) to replace cells lost to apoptosis, senescence, or therapeutic destruction of senescent cells; and by engineering the body to be impregnable to cancer (the OncoSENS WILT strategy).
In addition to damage to our main genetic code, the Hallmarks paper also puts mitochondrial DNA damage in the “genomic instability” Hallmark. We’ll return to this subject when we discuss the “mitochondrial dysfunction” Hallmark and its closest counterpart among the SENS Seven.

Epigenetic Alterations vs. OncoSENS
The Hallmarks paper classifies age-related epigenetic alterations as a primary Hallmark — that is, one of the (sources of) cellular damage. And certainly, some epigenetic alterations in aging cells are truly irreversible damage — i.e., “epimutations.” But cells also alter their epigenetic structures intentionally, as a way to turn different genes on or off to adapt to changes in their internal and external environment. One such important set of changes is, of course, cellular and molecular aging damage. In other words, “epigenetic alterations in aging” include some changes that really are primary Hallmarks (cellular damage in the form of epimutations), but others that are better considered as compensatory Hallmarks (ways that the cell adapts to aging damage).
And there’s a third reason why the epigenetic patterns in cells in a tissue can change with age: not changes in the epigenetic structures inside the same cells, but age-related shifts in the kinds of cells present in a tissue. For example, aging tissues often lose their workhorse cells to senescence or apoptosis, replaced by infiltrating fat cells (adipocytes). Aging tissues are also often infiltrated by immune cells, which work their way into the tissue to clear out senescent cells or damaged molecules.
The reason why changes in the kinds of cells present in a tissue can mislead scientists is that what makes one kind of cell different from another (say, the difference between a cell from the tubes that carry air into your lungs versus one of the airsac cells that extract the oxygen from that air) is which genes are turned on and turned off by the epigenetic structures that are specific to each cell type. So as the mix of cells in a tissue changes with age, the epigenetics of the cells in that tissue will change — but for a very different reason than either compensatory changes or epimutational damage occurring in the cells that were there in the first place.
It’s quite challenging to distinguish which of these three kinds of changes are occurring in aging tissues. SENS Research Foundation funded some of the early work on disentangling these changes, and scientists are still trying to work them out today.
Because each of these three distinct sources of epigenetic changes has a different mechanistic basis and different effects on our health from the others, putting them all into a single Hallmark and categorizing it as a primary Hallmark tends to muddy the water when one gets down to strategizing about longevity therapeutics to prevent and reverse them.
SENS, again, winnows these changes down to the underlying damage involved. SENS treats epimutations in a way that is operationally similar to how it deals with mutations (since both mutations and epimutations ultimately affect the cell by changing the products of genes). We can disarm those that send cells on the road toward cancer by WILT; we can eliminate those that lead to senescence by applying ApoptoSENS; and we can counteract those that lead to cell death through RepleniSENS.
There is one important difference between how SENS handles epigenetic aging changes versus damage to the genome. Unlike mutations in the DNA, a subset of epigenetic changes in aging tissues will likely be amenable to direct correction via epigenetic reprogramming. But harnessing this tool directly and widely to rejuvenate aging tissues is trickier and farther off than many of its most vocal proponents suggest. Instead, the clearest near-term use of reprogramming is to take a biopsy of an aging person’s cells and turn them into cells for transplantation. When done outside of the body, such cells can be screened for mutations or other problems, and when transplanted back into a person they will be plentiful, younger in some ways than the cells in the transplantation site, and will be accepted as fully “self” by the patient’s immune system.

Telomere Attrition vs. ApoptoSENS and OncoSENS
Telomeres certainly shorten with age in cells that divide, and that’s the focus of this Hallmark. Additionally, telomeres in aging heart muscle cells and possibly neurons and other long-lived nondividing cells suffer a more direct but non-shortening type of damage. This latter kind of damage had not yet been discovered when the original Hallmarks paper was written, but surprisingly, it wasn’t discussed in a recent update either.
While the “genomic instability” and “epigenetic alterations” Hallmarks have at least some overlap with the OncoSENS Strand of SENS, the difference between the two systems becomes stark when considering telomere attrition. When the Hallmarks paper talks about developing interventions against telomere attrition, the examples are of biotechnologically lengthening telomeres, even as the paper acknowledges that “There are several examples in which telomere attrition attenuates carcinogenesis [that is, protects against cancer] through limiting the replicative lifespan of malignant cells.”
In fact, this understates the issue: a mechanism for the cancer cell to maintain its telomeres is essential for a tumor to become life-threatening to a human, and is one of the original Hallmarks of Cancer. (If that sounds familiar, it’s because the Hallmarks of Cancer were the inspiration for the much later Hallmarks of Aging. The Hallmarks of Cancer stand on much stronger footing than the flattery-by-imitation Hallmarks of Aging, and are widely accepted as definitive descriptions of what a cell has to do to become cancerous).
At the same time that it acknowledges the risk of cancer from unrestricted telomere lengthening, the Hallmarks paper also spells out the harms that flow from telomere attrition. Critically, these harms are already covered by both the Hallmarks and the SENS Seven: “Telomere exhaustion explains the limited proliferative capacity of some types of in-vitro-cultured cells, the so-called replicative senescence, or Hayflick limit … DNA damage at telomeres is notably persistent and highly efficient in inducing senescence and/or apoptosis”.
Thus, the Hallmarks paper recognizes that telomere shortening is pathological precisely because of its downstream consequences on other Hallmarks rather than its independent effects — and that telomere lengthening carries yet another age-related risk (cancer). And yet it introduces telomere attrition as a separate Hallmark, despite having empaneled genomic instability, cellular senescence, and stem cell exhaustion as Hallmarks in their own rights.
If our purpose is to devise a list of targets for a comprehensive suite of longevity therapeutics, then setting up telomere attrition as a separate category of age change is ultimately redundant — and it suggests a solution (telomere lengthening) that is actively counterproductive. Instead, the SENS Seven includes and therapeutically targets each form of end-state damage that either telomere shortening or lengthening ultimately cause.
Thus, far from lengthening telomeres, denying cancerous cells the ability to do so is the core of both the WILT OncoSENS strategy and of 6-thio-2’-deoxyguanosine (6-thio-dG/thio) that has been suggested as a potential OncoSENS approach. Placing an absolute limit on cell replication will also likely prevent clonal haematopoieisis, another proliferative disease of aging, or at least make it easier to deal with using other strategies. And placing this lifetime cap on the division of abnormal cells can’t be done to cancerous cells without also doing it to cells that are not (yet) cancerous, because of the nature of cancer as a disease of runaway cell division with natural selection in its arsenal.
Placing this iron limit on cellular division will necessarily lead to more cells becoming senescent within the scope of our current lifespans. The damage-repair strategy is to allow this to happen, thus protecting the body from such cells’ cancerous potential — and then destroy them with senolytic strategies (ApoptoSENS), eliminating the twin threats of cancer and senescent cells. Alternatively, telomere attrition in such cells can trigger programmed cell death (apoptosis), which will even more definitively eliminate the risk of such a cell becoming cancerous. Either of these failsafe systems will also contribute to the loss of cells that our tissues need to function. The solution in that case is to allow such cells to fall on their swords — and preemptively replace them with cell therapy (RepleniSENS).

Loss of Proteostasis vs. LysoSENS
If our goal is to use these schemas as a way to strategically develop longevity therapeutics, there are two principal reasons to favor the SENS Seven category for intracellular aggregates (LysoSENS) over the “loss of proteostasis” Hallmark. These relate to how the loss of proteostasis Hallmark is conceptualized (and thus the implied solutions) and kinds of aging damage that it fails to capture.
“Proteostasis” refers to the ability of youthful, functioning cells to maintain a dynamic equilibrium in which its proteins assume and retain their proper 3-D structure. So loss of proteostasis means that the cell is failing to maintain this delicate balance and is instead accumulating misfolded, dysfunctional, excessive, and aggregated forms of those proteins. It takes a lot of machinery working properly to preserve proteostasis, as the manuscript makes clear:
All cells take advantage of an array of quality control mechanisms to preserve the stability and functionality of their proteomes. Proteostasis involves mechanisms for the stabilization of correctly folded proteins—most prominently, the heat-shock family of proteins—and mechanisms for the degradation of proteins by the proteasome or the lysosome. Moreover, there are regulators of age-related proteotoxicity, such as MOAG-4, that act through an alternative pathway distinct from molecular chaperones and proteases. [MR: There are many more players in proteostasis, such as osmolytes and the enzyme methionine sulfoxide reductase A (MsrA)]. All of these systems function in a coordinated fashion to restore the structure of misfolded polypeptides or to remove and degrade them completely, thus preventing the accumulation of damaged components and assuring the continuous renewal of intracellular proteins.
The first weakness of this Hallmark is one that it shares with most of the other Hallmarks as against the SENS Seven: that it describes a state into which an aging cell enters, rather than the bottom-line damage that the Hallmark causes or results from and thus a definitive target for therapy. By defining the problem as “loss of proteostasis,” the Hallmarks paper sends scientists into a chase for an implausibly wide and optimized proteostatic pharmacopoeia, with multiple drugs each intended to tweak some different component of the proteostasis-maintenance machinery.
And even in the most optimistic projection of such an enterprise, our inbuilt machinery would necessarily leave some fraction of the aggregates to accumulate and age-related disease to insinuate itself into our tissues, for the reasons common to all such “messing-with-metabolism” approaches that we delineated earlier.
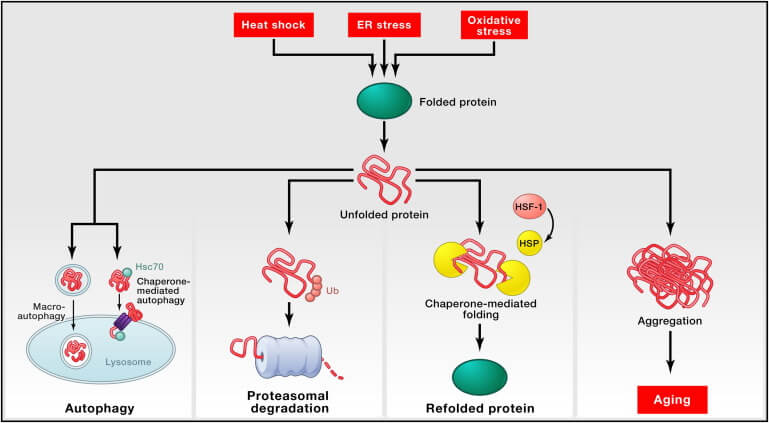
The SENS strategy takes a different tack. As the Hallmark paper clearly illustrates in this summary graphic, the point of all of this protein QC and waste management apparatus is to prevent the accumulation of intracellular aggregates — and it’s these aggregates that ultimately drive degenerative aging. So the damage-repair approach leaves all of this finely-tuned proteostatic machinery alone, and targets the aggregates themselves for direct destruction by novel enzymes and other LysoSENS rejuvenation biotechnologies.
So that’s the first knock on the “loss of proteostasis” Hallmark: that like other Hallmarks, it’s focused on process instead of product and thus mires us in excessive metabolic entanglement. The second is that it’s missing a significant amount of damaged material that accumulates inside aging cells.
The authors admitted to part of this in their 10-year update of the Hallmarks, in which they noted that “macroautophagy [which they had originally included as part of the proteostatic machinery] does not only affect proteins, but can target entire organelles and non-proteinaceous macromolecules.” Examples of such non-protein targets include damaged cholesterol products like 7-ketocholesterol that drive atherosclerosis, as well as lipofuscin (which is in significant part composed of the remains of damaged mitochondria and is rich in lipids in addition to proteins).
To cover over this breach in the Hallmarks, the authors added a new Hallmark in their ten-year update: “disabled macroautophagy.” (Similarly, a “forked” version of the Hallmarks produced during a symposium on the Hallmarks in Copenhagen proposed the addition of “compromised autophagy”). This would be a simple solution if the machinery for macroautophagy were neatly separated from the proteostasis machinery — but it’s not, which is why the original Hallmarks paper rightly included it as an important player in maintaining proteostasis. In addition to dealing with fatty molecules and some organelles, macroautophagy also sweeps up many proteins in the course of its Roomba-like patrol of the cell, and several distinct protein- and organelle-degrading systems that the Hallmarks paper reasonably classifies as part of the proteostasis machinery work by dragging specific damaged proteins or organelles to the same cellular “recycling center” (the lysosome) that is the end destination for macroautophagy.
Despite this, the followup paper insists on separating the two. In attempting to justify the artificial line, the authors tie themselves into knots as tangled as the stubborn bonds in lipofuscin:
Protein aggregates can also be removed by macroautophagy … Since autophagosomes [the “garbage bags” of macroautophagy] can envelop non-proteinaceous structures, this process will be discussed separately from proteostasis in the next hallmark section (disabled macroautophagy). Nonetheless, stimulation of autophagy constitutes a valid strategy for the elimination of intracellular protein aggregates.
When it’s working, the lysosome makes short work of many different kinds of damaged or past-due-date waste products — proteins, lipids, and organelles. When it’s not, the lysosome fails, macroautophagy stalls out, and the organelle can’t degrade any of them. So separating disabled macroautophagy from loss of proteostasis is ultimately arbitrary and unhelpful. The SENS Seven avoids this problem by focusing on what happens when any of these systems fail: the damage that accumulates inside cells and the biotechnology required to eliminate it (LysoSENS), regardless of how it originated or which elements of the cellular machinery failed to mitigate it in its earliest stages.

Mitochondrial Dysfunction vs. MitoSENS
You might think that mitochondria would be the one place where a Hallmark would finally overlap to the point of total eclipse with its cousin in the SENS Seven — but if so, you’d be surprisingly mistaken. In fact, this is arguably the one case where a Hallmark departs most completely from the seemingly-analogous member of the SENS Seven!
The MitoSENS category, like all the SENS Seven, is defined by accumulating damage to youthful biological structure: specifically, the accumulation of cells bearing mitochondria with DNA mutations, the most critical of which are large mitochondrial DNA deletions. It is these deletion-bearing mitochondria that completely take over cells in which they occur, and it is such cells that most clearly accumulate with age.
By contrast, the Hallmarks places mitochondrial DNA damage of all kinds into a corner closet of the “genomic instability” Hallmark. And when the Hallmarks paper says “mitochondrial dysfunction,” it instead refers to things that cause the mitochondria in an aging body to behave abnormally for reasons other than mitochondrial DNA damage.
To give the system its due, a significant fraction of the overall derangement of mitochondrial function in the aging body is the result of (mal)adaptations to — or consequences of — aging damage that occurs outside of the mitochondria themselves (and in many cases far away from the individual cells whose mitochondria are malfunctioning!), rather than the more direct effects of mitochondrial DNA damage. That’s why this Hallmark is classified as a “compensatory Hallmark” rather than a primary Hallmark.
For example, mitochondrial function becomes abnormal in many aging people because of insulin resistance, the interface of exercise, energy balance, and obesity in the muscles, and a different set of changes in the pancreas, despite the mitochondrial DNA being normal. It’s also abnormal in a variety of disease states (age-related and otherwise).
Mitochondrial function also declines in aging as a downstream consequence of a rising tide of DNA damage in the cell’s nucleus — including damage to genes that have nothing to do with the mitochondria. This is because of the dual functions of the enzyme AMPK in both catalyzing part of the DNA repair machinery and activating several aspects of mitochondrial function. As the nuclear DNA suffers increasing amounts of damage with age, AMPK is increasingly diverted away from the mitochondria to help repair the lesions. But with AMPK busy in its DNA repair role, it can’t turn on the proteins mitochondria need to function at their peak. As a result, aging mice suffer a decline in mitochondrial energy production, blunted mitochondrial replication, and greater fatigability during exercise.
Mitochondrial function is also impaired by the age-related decline in levels of the amino acid taurine, which is required by the “shuttles” that deliver amino acid building blocks to the cellular machines that produce the proteins that mitochondria use to generate cellular energy. Mitochondrial function is also abnormal in senescent cells for reasons unrelated to mitochondrial DNA damage (and that seem likely to instead relate to nuclear DNA damage). And while it remains an hypothesis, Dr. de Grey has speculated that aging cells may actively turn down the activity of their mitochondria in order to protect themselves from rising free radical damage. He got this idea in part from a study that found that administering mitochondrial activity-boosting dietary supplements to aging mice also increases mitochondrial free radical production, just as pressing harder on the accelerator of a car with engine wear leads to more tailpipe pollution.
This is why the Hallmarks paper categorized “mitochondrial dysfunction” as a “compensatory” rather than a “primary” Hallmark. While not all of these contributors to abnormal mitochondrial function in aging are “compensatory” in the sense of being actions the cell takes to offset the effects of primary aging damage, they are all the downstream results of damage to structures unrelated to the mitochondria.
By contrast, as we’ve already noted, the MitoSENS category is laser-focused on repairing the molecular damage to the mitochondria themselves, in the form of mutations — most importantly, large mitochondrial DNA deletions. Indeed, the MitoSENS lab at SENS Research Foundation is working to develop three different platform technologies (including the original MitoSENS strategy of allotopic expression) to replace or render harmless the mitochondrial DNA.
From the SENS perspective, all of the damage to cells and biomolecules outside the mitochondria that impinges on mitochondrial function is important, but it should be cataloged and repaired directly and for its own sake. Repairing such extra-mitochondrial damage would eliminate the “compensatory” changes that cause mitochondrial dysfunction in intact and otherwise-functional mitochondria. At that point, the mitochondria would be able to operate properly again. Conversely, addressing the many extra-mitochondrial drivers of the “mitochondrial dysfunction” Hallmark will fail if we don’t also develop a solution for mitochondrial mutations — and the Hallmarks offer none.

Cellular Senescence vs. ApoptoSENS
If “mitochondrial dysfunction” is the Hallmark farthest removed from its SENS Seven analog, then “cellular senescence” is the closest. They diverge on a theme we’ve seen repeatedly in comparing the two: the Hallmark focuses on the process of cells going senescent, while ApoptoSENS sets its sights squarely on the senescent cells themselves.
It’s worth noting that Hallmarks categorize “cellular senescence” as a compensatory Hallmark rather than a primary one. This is because cells undergo senescence as a response (“adaptation”) to conditions inside the cell that might make it harmful to the body — especially those that put it at risk of becoming cancerous. Because the Hallmarks paper is focused on the senescence process, and the process is protective, the authors of the Hallmarks paper are ambivalent about whether senescence could be targeted to combat aging (their “third criterion”):
We propose that cellular senescence is a beneficial compensatory response to damage that becomes deleterious and accelerates aging when tissues exhaust their regenerative capacity. Given these complexities, it is not possible to give a simple answer to the question of whether cell senescence fulfills the third ideal criteria for the definition of a hallmark.
Some of their caution is explained by the fact that they were writing in 2013, when scientists had only ever tested a senolytic-type intervention in mutant mice. Thus, they didn’t yet have evidence that destroying senescent cells protects otherwise-healthy aging mice against many aspects of age-related decline. Nor did we yet have rationally-designed drugs that execute senescent cells and rapidly rejuvenate old lab animals. But with or without this evidence, so long as you focus on the senescence process rather than on targeting the senescent cells themselves, you will always be ambivalent about interfering with it.
By contrast, Dr. de Grey already had the foresight in the original 2002 paper outlining the SENS approach that the biomedical solution to cellular senescence was to destroy the senescent cells after they arise. He came to this strategy long before there was direct experimental evidence supporting it because of the core logic of the “damage-repair” approach. While the senescence process may be adaptive, the accumulation of senescent cells in the aging body constitutes aging damage — and it’s damage to our cells and biomolecules that compromises tissue function, robs us of resilience, and degrades our health as we age. The solution was therefore to allow cells to turn senescent as they “need” to — and then liquidate them before they can accumulate to harmful levels. An avalanche of animal evidence and limited human data have now validated that strategy.
An additional limitation to the Hallmarks is that there is no category for the accumulation of abnormal cells other than senescent ones, such as T-cells that attack the “nurseries” for new neurons or that infiltrate fat tissue and contribute to insulin resistance with age. ApoptoSENS very deliberately includes eliminating all cells that have outstayed their welcome.

Stem Cell Exhaustion vs. RepleniSENS
Both the Hallmarks and the SENS Seven recognize that declining numbers and function of stem cells contribute to degenerative aging. But there are two problems with framing the challenge as “stem cell exhaustion.”
The first problem is that the authors seem to focus entirely on the depletion (and to a lesser extent, dysfunction) of the body’s existing tissue stem cell pool — and if you think of the problem that way, there’s no clear way to do better than evolution has already equipped us to do.
We are born with a limited number of stem cells, so for them to last a lifetime, they need a way to keep their telomeres long. The body already tries to do this by equipping tissue stem cells with the telomerase enzyme, which could in theory allow them to keep reproducing themselves indefinitely. Additionally, stem cells usually guard their “stemness” through a process called asymmetric cell division, in which the “parent” stem cell divides to create two quite different cells. One of the original stem cell’s daughter cells becomes a specialized cell needed by the injured or aging-worn tissue (like a blood stem cell or a brain neuron), while the other remains a tissue stem cell like its parent, allowing the stem cell line to continue.
If this system worked perfectly, then our tissue stem cells would keep producing new cells to support their target tissues indefinitely, as there would always be viable stem cells left over after sending out a new specialized cell for maintenance and repair. But despite having these mechanisms to maintain the stem cell pool, the body’s supply of tissue stem cells clearly dwindles with age, and those stem cells that remain become less effective at doing their job.
Some of the loss of stem cell function with age is due to suppressive signals in their environment rather than because of defects in the stem cells themselves. For instance, the age-related stiffening of the tissue in which muscle stem cells are embedded impairs their ability to reproduce and mature. Similarly, scientists have restored much of the flagging ability of muscle stem cells to repair muscle injuries by diluting out the suppressive signals in the local environment via heterochronic parabiosis or therapeutic plasma exchange — or at least, they have done so in late middle-aged (20-month-old) mice. But in truly old mice (28 months), the stem cells themselves become damaged enough that this rejuvenating effect dwindles away to nothing, demonstrating the need to repair the damage in aging tissue stem cells or replace them entirely.
But obviously, changes in the stem cells’ environment can’t bring back stem cells that have been lost entirely, and it’s not clear what you could usefully do about that. Scientists have given extra copies of the telomerase gene to keep their tissues proliferating into more advanced ages, but we’ve already discussed the dilemma posed by telomerase. It’s no coincidence that cancer arises mostly in cell types that already proliferate (as opposed to cells like muscle cells and neurons that are produced once and last a lifetime), and that it’s especially likely to rear its head in stem cells. And there’s a similar dilemma in “deciding” when to allow a tissue stem cell to replicate itself and when to enforce quiescence (that is, keeping the stem cell dormant and not reproducing itself or producing new tissue cells). The Hallmarks paper lays out the dilemma nicely (all emphasis mine):
Although deficient proliferation of stem and progenitor cells is obviously detrimental for the long-term maintenance of the organism [because it fails to replace the cells a tissue loses with age], an excessive proliferation of stem and progenitor cells can also be deleterious by accelerating the exhaustion of stem cell niches. The importance of stem cell quiescence for the long-term functionality of stem cells has been compellingly demonstrated in the case of Drosophila intestinal stem cells, in which excessive proliferation leads to exhaustion and premature aging …
The authors then offer a few examples of therapies that have worked to counteract these problems in mice:
recent studies have shown that an increase in FGF2 signaling in the aged muscle stem cell niche results in the loss of quiescence and eventually in stem cell depletion and diminished regenerative capacity, whereas suppression of this signaling pathway rescues these defects. This opens the possibility of designing strategies aimed at inhibiting FGF2 signaling to reduce stem cell exhaustion during aging. Along these lines, [Calorie restriction] has been reported to increase intestinal and muscle stem functions … mTORC1 inhibition with rapamycin … may also improve stem cell function in the epidermis, in the hematopoietic system, and in the intestine …
Each of these therapies puts a thumb on the quiescence-proliferation seesaw in favor of quiescence and thereby prevents stem cell exhaustion. But most of the examples they give are based on short-term studies that can’t tell us whether they come at the cost of less cell proliferation and repair. On the other hand, we know that this is exactly what happens in the case of Calorie restriction, which preserves the ability of cells to proliferate into old age — but at the cost of slower cell proliferation and impaired wound healing in the meantime. And the CR animals can only tap into that saved-up proliferative capacity if scientists refeed them to allow their cells to replicate themselves again.
The other problem with framing the problem as “stem cell exhaustion” is that there are plenty of tissues in the body that either have no resident stem cells to exhaust, or that have far too few such cells to sustain tissues into what are currently advanced ages even you could keep their youthful numbers and functions at their peak. For example, scientists debate how much the adult human brain is able to generate new neurons (neurogenesis), but even under the most optimistic reading of the evidence, new neurons are birthed in and integrate into only a tiny fraction of the brain: they do nothing to replace neurons that are lost to aging or trauma outside of these very restricted zones.
Similarly, while the adult heart has some capacity to generate new heart muscle cells, the replacement rate is only 1% per year in 25-year-olds. The replacement rate for these cells falls to 0.3% annually by the age of 75, so you can imagine an anti-stem-cell-exhaustion treatment to combat this decline. But even if we developed a therapy that would preserve the numbers and functions of the relevant cardiac stem cells at the peak levels of a 25-year-old, replacing only 1% of the cells in the heart each year would by definition take 100 years to complete the cycle. As it stands, we lose about a third of our heart muscle cells between ages 17 and 90 — and that’s in people who don’t have heart failure and who have never suffered a heart attack or trauma, any of which increases the need for new heart muscle cells and potentially destroys the cell population responsible for doing the replacement.
So even if we could completely prevent “stem cell exhaustion” and keep our bodies’ tissue stem cell pools topped up and functioning as well as they do in the prime of our current lives, they would still be unable to keep all of our tissues supplied with the cells they need to keep us alive and healthy.
Instead of defining the problem as “stem cell exhaustion,” RepleniSENS squarely focuses on the loss of all kinds of cells to aging — including both stem cells and mature cells from all tissues. And the therapeutic strategy is not to ask our existing, inborn tissue stem cells to do something they were never designed to do, but to replace lost cells and tissues directly using cell therapy and tissue engineering.
The last thing to note about this Hallmark is that the paper classifies it as an “integrative” Hallmark: that is, one of the two Hallmarks that they say “arise when the accumulated damage caused by the primary and antagonistic hallmarks cannot be compensated by tissue homeostatic mechanisms”, “directly affect tissue homeostasis and function”, “and likely constitutes one of the ultimate culprits of tissue and organismal aging.”
But of course, the body does adapt in a multitude of ways to carry on function in the face of cell loss. For example, the more prominent symptoms of neurodegenerative aging of the Parkinson’s type are caused by the loss of specialized neurons in a specific area of the brain that produce the signaling molecule dopamine. As these neurons are lost, the surviving dopamine-producing and dopamine-using cells change their metabolism to maintain the level of dopamine available for their target cells. This allows a person to carry on functioning even as he or she loses more and more of these critical neurons, only to fail rapidly when cell loss exceeds 85%.
More remarkably, as people with diabetes burn out the insulin-producing cells in the pancreas, other cells in the organ that do not produce insulin appear to pick up the slack by transforming themselves into insulin-producing cells.
More generally, all the Hallmarks are “integrative” in the sense that they cause dysfunction and are both produced by and feed forward into other kinds of aging damage. This point is on more open display in the authors’ tenth-anniversary lookback on the Hallmarks, in which illustrations consistently highlight the functional consequences of damage or disruption to each Hallmark.
Missing in Action
Up until now, we’ve been discussing Hallmarks and members of the SENS Seven that have some level of overlap, and have contrasted the ways that the Hallmarks and the corresponding members of the SENS Seven frame the biology of aging, the point in the aging process in which they ground each category (ongoing processes vs. accumulating damage), and how they inform strategies for developing longevity therapeutics. But there are two particular kinds of aging damage that the Hallmarks ignore entirely. If we are to conquer aging, we can’t afford these blind spots.

AmyloSENS
As we discussed above, the original Hallmarks paper failed to account for non-protein waste products inside the cell in the “loss of proteostasis” Hallmark, and the ten-year update on the Hallmarks only addressed this blind spot by way of the non-solution of offloading them arbitrarily into a new “disabled macroautophagy” Hallmark. But the original Hallmarks paper took no account at all of the accumulation of damaged molecules outside of the cell,* and the update paper only mentions them as one consequence of the loss of proteostasis inside the cell (which is in any case only one source of extracellular aggregates).
Even if all the extracellular aggregates were the downstream results of proteostatic failure (and again: they’re not!), it would be a bad therapeutic strategy to focus entirely on fine-tuning proteostasis and simply ignore such aggregates once they are formed. And what makes this omission so glaring is that “AmyloSENS” molecular damage includes some of the most well-established drivers of age-related disease and debility, such as beta-amyloid (the chief driver of neurodegenerative aging of the Alzheimer’s type) and cardiac TTR amyloid (an important contributor to heart failure and likely to declining heart function with age in all aging people), as well as the extracellular versions of tau and alpha-synuclein aggregates and IAPP aggregates in diabetes.
In fact, rejuvenation biotechnologies targeting extracellular aggregates are now on more solid footing than any other class of longevity therapeutic. AmyloSENS antibodies targeting beta-amyloid oligomers and protofibrils are the first “damage-repair” longevity therapeutics to be approved by FDA, following Phase III clinical trials that showed them to be the first therapies of any kind that can put the brakes on Alzheimer’s neurodegeneration. Other AmyloSENS antibodies targeting cardiac TTR appear poised to reverse an important cause of heart failure. And yet more therapies directly targeting other extracellular aggregates are in the pipeline. Any blueprint for a “divide-and-conquer” strategy against degenerative aging needs to include these aggregates and a way to eliminate them. The SENS Seven do; the Hallmarks don’t.

GlycoSENS
A very large amount of age-related disease, disability, pain, and death is the direct and in-your-face-obvious result of damage to the extracellular matrix (ECM) — that is, the intricate lattice of structural proteins that provide support, structure, and signaling to our cells. When people talk about how aging has afflicted them, their top-of-mind examples are often pains in their backs, joints, and tendons. Meanwhile, age-related weakening of the bones (osteoporosis) is a major cause of death and disability in older people. And on top of these are the many plagues of the aging body that are caused by ECM damage but that are not directly perceived by a person, such as the stiffening of the large arteries that leads to strokes and kidney damage.
So it’s perplexing that the original Hallmarks paper neglected damage to the ECM — and even more perplexing that the tenth-anniversary paper jammed it into the “altered intercellular communication” Hallmark!
It’s true that damage to functional molecules in some kinds of ECM distorts the signaling that intact ECM molecules provide to the cells they support. For instance, age-related stiffening of the ECM that supports muscle stem cells impairs their ability to proliferate and mature. But to ignore the more obvious and potentially catastrophic ways that damaged ECM can directly limit our motion or end our lives would be like writing a report about the health impact of smoking that focuses on its effect on cardiovascular disease while being silent on lung cancer.
And it makes even less sense to confine such damage to the “altered intercellular communication” Hallmark when you consider that (a) much of the damage suffered by aging ECM is inflicted by non-biological processes like UV damage to the skin; (b) much of the rest of the damage is wrought by mechanical forces, such as the cyclic stress of the pulse pounding against the elastin structures in the artery wall; and (c) to the extent that biochemical processes contribute to damaging ECM, the main offenders are not signaling molecules but enzymes that either directly break it down or that are abnormally activated and synthesize too much of it.
These two cases — the incongruous shoehorning of ECM damage into the “altered intercellular communication” Hallmark and the relegation of extracellular aggregates to an aside in the discussion of “loss of proteostasis” — may in part be the result of the Hallmarks being so focused on damage to cells as the driver of aging. You can see this in the way the authors framed their process in the original paper: “The time-dependent accumulation of cellular damage is widely considered to be the general cause of aging (Gems and Partridge, 2013; Kirkwood, 2005; Vijg and Campisi, 2008). … [C]ancer and aging can be regarded as two different manifestations of the same underlying process—namely, the accumulation of cellular damage.” But while that may help explain these near-omissions, they don’t bring the Hallmarks up to the task.
The Copenhagen “New Hallmarks of Ageing” proposed a new “alt-Hallmark” for “altered mechanical properties” that doesn’t quite fit the bill either. This proposed Hallmark includes ECM damage but also changes in mechanical properties inside the cell. These intracellular mechanical properties are quite different from ECM damage in their origin, in their effects on the body, and in how we can target them. Many of them are also clearly captured elsewhere in the SENS Seven. Take, for example, the destabilization of proteins inside the envelope surrounding the nucleus that happens during cellular senescence. As with other changes in senescent cells, there’s no need to separately target this destabilization since we are going to destroy senescent cells anyway.
If our goal is to bring degenerative aging under full medical control, then we need to give ECM damage the priority it deserves as a key target in a framework for longevity therapeutics. Ten years before the original Hallmarks paper and twenty years before the update, ECM damage and rejuvenation biotechnology strategies to repair it were incorporated as the GlycoSENS strand in the SENS Seven. Thanks to that early focus, SENS Research Foundation-funded research has spawned a spinoff to break the most prominent crosslink in aging collagen, and another longevity biotech startup is pursuing a strategy to repair frayed and calcium-stiffened elastin in our arteries.


The Leftovers
While the Hallmarks glaringly lack more than distant echoes of two key members of the SENS Seven, the SENS Seven omit two Hallmarks because they will be redundant once the SENS Seven are removed or repaired by rejuvenation biotechnologies.
We’ve already discussed the “altered intercellular communication” Hallmark in some detail as part of critiquing the “compensatory/antagonistic” subgroup of the Hallmarks. The Hallmarks paper classifies altered intercellular communication as an “integrative” Hallmark (i.e., one that “arise[s] when the accumulated damage caused by the primary and antagonistic hallmarks cannot be compensated by tissue homeostatic mechanisms”). But nearly all changes in intercellular communication in aging are actually “compensatory” in nature: ways the body tries to maintain function in the face of “primary Hallmarks” (cellular and molecular aging damage (or damaging processes)). We illustrated this with several specific examples in the discussion of the integrative Hallmarks above, and could give many more examples.
SENS sweeps these compensatory signaling changes away by going directly after the damage itself. Meanwhile, the few cases of altered intercellular communication that are not compensatory are the direct results of damaged cells and biomolecules not doing their job or doing it abnormally, such as the SASP from senescent cells or deranged signaling generated by damaged ECM. With the damage repaired, cellular signaling will return to normal, just as inflammation resolves after a wound is healed.
The last Hallmark to address is “deregulated nutrient sensing.” This Hallmark doesn’t quite know if it is coming or going. On the one hand, the best-studied old-school geroscience anti-aging interventions are all ways to inhibit nutrient signaling of one kind or another: Calorie restriction and inhibiting the mTOR pathway or insulin/IGF-1 signaling (IIS). On the other hand, these same nutrient signaling pathways themselves go awry in aging — and often in confounding ways, as the Hallmarks paper lays out:
Paradoxically, GH and IGF-1 levels decline during normal aging, as well as in mouse models of premature aging. Thus, a decreased IIS is a common characteristic of both physiological and accelerated aging, whereas a constitutively decreased IIS extends longevity. These apparently contradictory observations can be accommodated under a unifying model by which IIS downmodulation reflects a defensive response aimed at minimizing cell growth and metabolism in the context of systemic damage (Garinis et al., 2008). According to this view, organisms with a constitutively decreased IIS can survive longer because they have lower rates of cell growth and metabolism and, hence, lower rates of cellular damage. Along the same lines, physiologically or pathologically aged organisms decrease IIS in an attempt to extend their lifespan.
On the one hand, the defensive model the authors lay out above doesn’t quite hang together. First, inhibiting IIS is only effective to any meaningful degree when it is initiated during development and youth and has very little benefit when begun late in life. And conversely, raising old rodents’ IGF-1 levels back to their youthful norm protects them against some of the harmful impacts of aging. So a rheostat to turn down an organism’s IGF-1 levels when it is already in late-life decline doesn’t make sense even in principle.
And postulating the existence of such a rheostat stands in stark contrast with the fact that most studies find that mTORC1 is hyperactivated in aging tissues. If the body were turning down IGF-1 to protect itself from that hormone mediator’s harmful effects, why would it simultaneously jack up mTOR activity — especially when (unlike IIS) inhibiting mTOR late in life with rapamycin does exert anti-aging effects?
And even if turning down IGF-1 late in life did protect old mice from aging damage, it doesn’t make sense from an evolutionary standpoint that such a rheostat would exist. There is no evolutionary pressure to develop systems to slow down aging (or do anything else) if they only kick in late in life. Indeed, it’s exactly because of natural selection’s blindness to events that impact survival later than when an organism typically dies in the wild that organisms don’t evolve the machinery to live longer than they do in the first place.
But the key point on which longevity therapeutics development needs to focus is buried in that same section. These pathways impact aging because they achieve “lower rates of cell growth and metabolism and, hence, lower rates of cellular damage.” If we remove and repair that damage with rejuvenation biotechnologies, we will have no need to concern ourselves with interfering with the processes that lay it down in the first place.
(Before leaving this topic, I will note that in their discussion of the “impaired nutrient sensing” Hallmark, both of the Hallmarks papers endorse the idea that SIRT1 is an important node in the anti-aging effects of Calorie restriction. With the notable exception of SIRT6, the evidence that sirtuins (and particularly SIRT1) are important players in aging in general or in mediating the effects of CR has largely fallen apart).
No Matter How You Slice It, We Need to End Aging
For a decade after Dr. de Grey and colleagues released the first version of the SENS Seven, most scientists working in aging were skeptical about whether the divide-and-conquer, damage-repair approach that the paper introduced was feasible. One of the great services that the Hallmarks of Aging have rendered is to make that idea mainstream. As we’ve seen, the SENS Seven lays out the problem in a more strategic way, such that we can be confident that developing a comprehensive suite of rejuvenation biotechnologies that remove, repair, or replace the damage they catalog is a more efficient and exhaustive path to the medical conquest of aging. At SENS Research Foundation, we work every day to implement it.
* The original Hallmarks paper makes one reference to “amyloid-binding components” as part of the proteostatic machinery, which you might think is intended to incorporate extracellular aggregates. But the compounds in question target aggregates inside the cell, through both direct action on the amyloidogenic proteins and by stimulating the intracellullar proteostatic machinery.