
With the enormous contemporary economic and human costs of Alzheimer’s disease (AD) and other dementias of aging, and with the global prevalence of AD projected to quadruple by 2050,(1) disease-modifying therapies for AD are desperately needed. A key target for rejuvenation biotechnologies to prevent and arrest the course of AD is the removal of aggregated beta-amyloid protein (Aβ) from the brain. Amongst the constellation of factors playing some role in the pathogenesis of AD, there is now a strong case for the thesis that Aβ aggregates are at the root of its aetiology.(2,3) Moreover, Aβ has also been implicated in the cognitive deficits clinically present in other age-related neurological diseases. For example, brain Aβ deposits and abnormally low levels of Aβ42 in the cerebrospinal fluid (CSF) discriminate people suffering from Parkinson’s disease dementia and dementia with Lewy bodies from those suffering from Parkinson’s disease but whose cognition remains intact.(20-23) And even amongst people whose cognition is still within the normal range, the presence of Aβ plaque as detected on [11C] Pittsburgh compound B (PiB)-Positron Emission Tomography (PET) is associated with cognitive deficits and increased rate and extent of gray mattter atrophy.(37)
The need for disease-modifying therapies in AD, and the strength of the case for Aβ as a target, have recently driven substantial regulatory reform and innovations in clinical trial design to acommodate more effective testing of Aβ immunotherapies targeting the removal of Aβ from the brain. The first fruits of these changes are the initiation of a series of large, late-stage clinical trials of Aβ vaccines in the early clinical or even preclinical phases of the disease. These reforms have wider and even more hopeful implications, because they open up the path for faster and more meaningful clinical trials of other rejuvenation biotechnologies as they emerge.
Charting an EXPEDITION
On July 19, pharmaceutical company Eli Lilly announced the launching of “a new pivotal trial” for solanezumab, their passive vaccine targeting Aβ monomers (and possibly small-n oligomers). As with most other regemerative therapies for the removal of extracellular aggregates, solanezumab aims to remove existing accumulations of Aβ from the brain by binding to the peptide and mobilizing it from the brain, whether by acting from within the brain itself, or indirectly by binding to Aβ pools located in “peripheral sinks” and thereby altering the dynamic equilibrium between central and peripheral pools.(4,15) The trial, to be named EXPEDITION-3, will incorporate lessons learned from the prior EXPEDITION-1 and -2 solanezumab trials. Those earlier trials randomized 2,052 subjects with diagnosed AD to receive 400 mg of intravenous solanezumab or placebo once every 4 weeks for 80 weeks, and included subjects with a range of severity of cognitive impairment from the “mild” to the so-called “moderate” (Mini-Mental State Examination (MMSE) score of 16-26 at screening).
Lilly originally reported that when the primary analysis of EXPEDITION-1 and -2 was performed, including all enrolled subjects in an intention-to-treat model, no benefit of treatment was evident. By contrast, both Lilly’s own prespecified secondary analysis of data pooled from both trials(5) and an independent analysis of the raw study data performed by the National Institute on Aging (NIA)’s Alzheimer’s Disease Cooperative Study (ADCS) group(6,7) indicate a substantial disease-modifying effect in subjects with mild (and thus, likely early-stage) AD (MMSE 20-26 at screening).(8)
To maximize the robustness of their conclusions, ADCS established three independent study analysis groups, each of which applied their own process of assignment, imputation of missing data, and analysis to the raw data provided by Lilly. After performing multiple sensitivity analyses, and applying the most conservative statistical analysis to the results, the ADCS group concluded that “a very coherent picture … emerges, and that is that there is a cognitive benefit.”(7) In their analyses, the effect of treatment on cognition in the trial population as a whole ranged from a 15 to a 20% deceleration in the rate of cognitive decline on different tests of cognitive impairment, although most of those results did not reach statistical significance. Importatantly, however, this effect was clearer and stronger in the subgroup with mild/early AD.
In participants with mild AD, solanezumab reduced the rate of cogntive decline by 34% (30-35% in different analyses) as assessed on the primary cogntitive assessment scale (the Alzheimer’s Disease Assessment Scale cognitive subscale (ADAS-Cog 14)); it also decelerated cognitive decline by a similar amount on the MMSE, and afforded smaller but significant benefits on most other cognitive scales.(7,8) The primary result was highly robust against variations in analysis, and in most sensitivity analyses the statistical significance of the results increased. The ADCS analysts also found that solanezumab decelerated functional decline by 18% in mild/early AD subjects, as assessed on the Alzheimer’s Disease Cooperative Study Instrumental Activities of Daily Living inventory subscale (ADCS-IADL) (P=0.045), and on the ADCS-Activities of Daily Living (ADCS-ADL) scale as well.(7) On the basis these results, combined with Lilly’s own analysis of the data from the mild vs. moderate AD subgroups of the earlier trials, EXPEDITION-3 will recruit exclusively subjects with mild AD.

Figure 1. Labeling of Brain Aβ Deposits with 18F-florbetapir. Reproduced from (44) with permission. © 2012 Society of Nuclear Medicine and Molecular Imaging (SNMMI).
There was also some suggestion of a reduction in brain Aβ deposits in solanezumab-treated volunteers with mild AD, as measured using the PET tracer 18F-florbetapir.(7,8) This result was at P=0.10, but the lack of statistical significance may reflect underpowering rather than statistical noise, because only a subset of volunteers underwent florbetapir-PET. Additionally, there was a substantial increase in total CSF Aβ42 and -40 in treated subjects, and a dose-dependent increase in CSF free Aβ42 in subjects administered 400 mg/d.(11) In addition to indicating target engagement by solanezumab, the rise in CSF Aβ42 is consistent with a postitive therapeutic effect, since CSF Aβ42 is consistently observed to decline during the period leading in to AD diagnosis.
There is an additional aspect of the PET and CSF biomarker data that suggests that the original EXPEDITION trials may have diluted the potential benefit of solanezumab because of inaccurate patient diagnosis and recruitment. The volunteers recruited for those trials had been diagnosed with AD based entirely on clinical criteria, but fluorbetapir-PET testing of a subset of participants revealed that 26% of tested subjects had no amyloid pathology.(7,8) This suggests that many of the subjects’ cognitive impairments were not driven by Aβ, but by other causes such as frontotemporal or vascular dementia. In such subjects, any effect of an Aβ-targeting therapy would be be expected to be much more limited than in subjects with AD as a disease of Aβ, suggesting that the true treatment benefit in subjects with (mild) AD may have been greater than was reported. As a result, EXPEDITION-3 will recruit only subjects with an AD diagnosis confirmed to be amyloid-associated with PET or CSF data.
Finally, EXPEDITION-3 will recruit an estimated 2,100 subjects — nearly double the number of people with mild AD recruited into the original trials.
CAPping the Pyramid
The announcement of EXPEDITION-3 was very good news for the advancement of rejuvenation biotechnology — and it follows a series of previous announcements of independent trials testing Aβ-targeting and other regenerative therapies in presymptomatic or early AD. Three of these are now coordinating in a consortium known as the Collaboration for Alzheimer’s Prevention (CAP), which exists to facilitate communication, avoid redundancy, ensure that data are collected and presented in ways that make the different trials’ results maximally comparable, and mutually negotiate the regulatory hurdles facing preventive AD trials.
While the focus on mild AD in EXPEDITION-3 is justified by prior results with solanezumab, a key feature of the CAP trials is that they include significant numbers of people who do not yet have AD — people who, in some cases, show no cognitive deficits at all. That such trials would be allowed at all is the output of a recent and urgently-negotiatiated series of stakeholder meetings amongst scientists, AD patient advocacy organizations, and FDA,((12); see also here and here) which have established precedents that will be critical to future clinical testing of these and future rejuvenation biotechnologies.(42,43) Fortunately, solanezumab was found to be quite safe in EXPEDITION-1 and -2: few treatment-associated adverse events ocurred, and many of these were attributable to lumbar puncture for CSF biomarker collection rather than treatment. In contrast to some other Aβ-targeting vaccines, vasogenic oedema did not emerge as a major concern.(11) This will doubtless have eased the minds of Eli Lilly, the US Food and Drug Administration (FDA), and institutional review boards in taking the very unusual step of approving large clinical trials in volunteers who are not yet suffering with a “disease” indication.
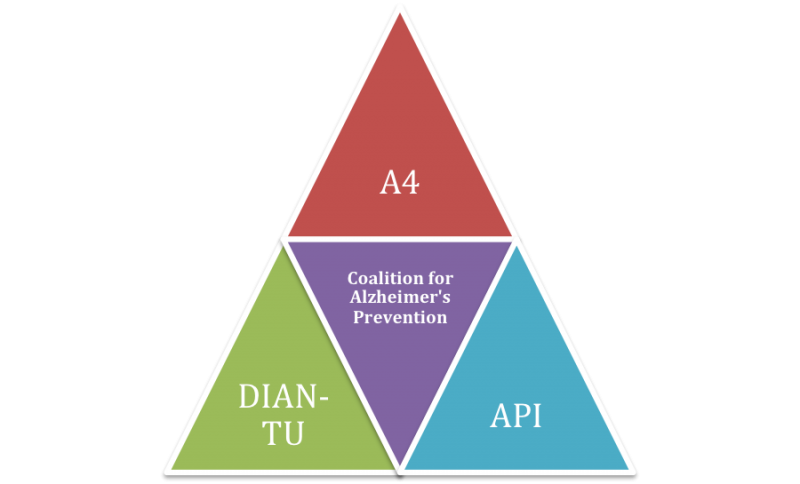
Figure 2. CAP coordinates several preventitve and early-stage AD trials using Aβ immunotherapies.
DIAN-TU is an intervention arm of DIAN, a highly productive observational study tracking the adult children of persons with known determinative AD mutations. The study includes both mutation carriers and non-carriers from these families, in pre-symptomatic and symptomatic stages of the disease, and tracks the course of dominantly-inherited AD starting at a point when many participants are still cognitively normal, following them longitudinally through the trajectory of AD. DIAN has used CSF biomarkers, PiB-PET for amyloid, and clinical and cognitive tests to map out the trajectory of the inherited disease and compare it to AD resulting from “normal” aging alone.
Like DIAN, DIAN-TU will recruit volunteers whose parents succumbed to AD as a result of determinative mutations. Unlike DIAN, DIAN-TU trial participants will be no more advanced than mild AD (to include cognitively normal and MCI subjects), and will be as much as 15 years younger — and no more than to 10 years older — than their mutation-bearing parent’s age of symptom onset. Volunteers with mutations (n=160) will receive either 400 mg IV solanezumab, 225 mg subcutaneous gantenerumab (an Aβ vaccine that we have covered before), or placebo; 80 noncarriers will be assigned blindly to the placebo group, to preserve blinding as to both mutation status and intervention, while avoiding exposing subjects who are not at near-term risk of AD to experimental therapies. A previously-announced beta-secretase inhibitor arm was evidently removed from the most recent posting of the DIAN-TU protocol, presumably following Lilly’s halting of Phase 2 trials with the drug (LY2886721) due to hepatotoxicity. The trial outcomes will focus on PET-detected amyloid, CSF Aβ and tau, rate of brain atrophy on volumetric MRI, and change in 2-[18F]fluoro-2-deoxy-D-glucose (2-DG) PET metabolism in specific regions of interest; cognitive measures will be included in exploratory analyses.
The API is similar to DIAN-TU, in that it is an interventive extension of an existing longitudinal study of biomarkers and cognition in persons afflicted with a determinative AD mutation. In this case, volunteers for the trial (n≈300) will be selected from members of a kindred in Medellin, Colombia that can be traced to a single common ancestor, and all participants will be carriers or noncarriers of the same presenilin-1 mutation. Carriers typically become clinically demented at approximately age 45. Approximately 24 participants from the United States will also be included. Asymptomatic mutation carriers will receive either monthly injections of the immunotherapy (crenezumab) or placebo (100 of each); similarly to DIAN, an additional 100 noncarriers will also receive placebo. The API is spearheaded by the Banner Alzheimer’s Institute, in collaboration with Dr. Francisco Lopera and others at the University of Antioquia in Colombia; the NIH, which is providing the bulk of the funding; and Genentech, which is supplying some of the funding and the study antibody.
The A4 trial, facilitated by the ADCS, is perhaps the most aggressively preventive of the CAP trials: it will enroll 1000 persons aged 65 and older, with normal cognitive function and no specific genetic risk for AD, but with evidence of amyloid deposition on PET screening. Much of the funding will come from NIA, with additional support from the private sector. Subjects will be treated for three years with solanezumab or placebo, with an intended post-trial followup study. (See additional details reported here and here).
Experimental Variation with Scientific Selection
The collaborative establishment of similar assays and reporting methods in the CAP trials has the advantage that it may highlight the relative strengths and risks of the three Aβ immunotherapies being tested across the trials. Notably, each of these vaccines appears to remove Aβ from the brain through a distinct mechanism of action (see Figure 3). Solanezumab appears to engage monomeric Aβ in the periphery and effect a drawdown of CNS Aβ via the “peripheral sink” mechanism, along with some possible CNS binding of monomers (and possibly small-n oligomers) and prevention of higher-n assembly. Crenezumab appears to have a wide range of affinities, binding to oligomeric and fibrillar forms of Aβ with high affinity while still capturing some monomeric Aβ. And gantenerumab evidently binds to both N-terminus and mid-sequence Aβ epitopes, with a strong affinity for fibrillar species; human and animal studies suggest that it opsonizes brain Aβ, recruiting brain microglia for phagocytosis for clearance.(13,14)
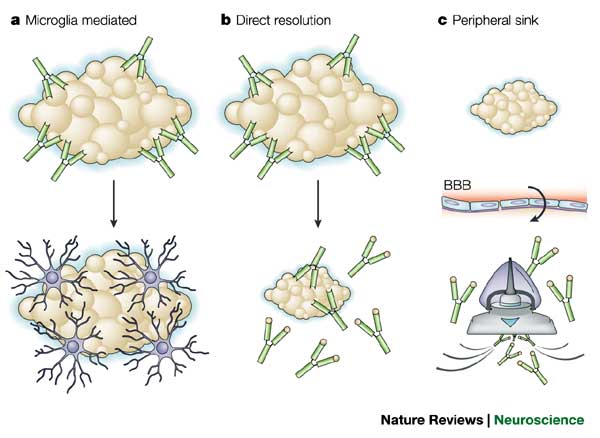
Figure 3. Mechanisms of action of different Aβ immunotherapies exploiting the acquired immunity paradigm. ©Nature Publications Group. Reproduced from (15) with permission.
Similarly, the fact that the volunteers in these trials will begin treatment at different points along the trajectory to dementia, but with none of them more advanced than mild AD, will give an important signal on the effectiveness of an Aβ-only approach across a spectrum from prevention to treatment. In fact, by the time the CAP trials are complete, we will have data from at least one additional stage along this continuum: Hoffman-La Roche is currently recruiting subjects with prodromal AD for a phase 2/3 trial they are designating SCarlet RoAD. In this trial, “prodromal AD” will be defined as subjects with mild cognitive impairment (MCI) (MMSE>24) and CSF Aβ and tau levels consistent with Aβ-driven disease rather than another early-stage dementia. SCarlet RoAD will test the ability of gantenerumab to slow or arrest decline in cognitive and functional ability in these sujects (as assessed on Clinical Dementia Rating scale Sum of Boxes (CDR-SOB)), and its effects on the change in brain amyloid as measured with PET (which ligand is not specified in the ClinicalTrials.gov registration).
The Needle and the Damage Not Undone
These multiple large-scale, randomized controlled trials in people whose clinical status ranges from within the normal range up through early/mild AD crystalizes the emerging view in the AD research community that with the long time-course of the cascade of etiopathogenic events driving the emergence of dementia, successful prevention and treatment of AD with Aβ-targeting therapies will require early — possibly even presymptomatic — intervention. One might in fact question whether even these trial designs intervene early enough if their sole target is Aβ (and all the more so since, in each case, a single vaccine with affinity for a limited range of Aβ assembly forms is being used).
The ultimate diagnosis of dementia of the AD type is the outcome of a long and mostly clinically silent period of insidious age-related accumulation of cellular and molecular lesions in the CNS. Evidence from the observational arms of DIAN and API(17-19) in carriers of determinative AD mutations, and subsequent studies in the general population in the United States(38) and Australia,(39) have have sketched a detailed picture of the trajectory of the key structural and functional changes leading into clinical AD in human patients. In the general population, Aβ begins to accumulate in the brain approximately 15-20 years before the first definitive appearance of plaque on PiB-PET, and this in turn occurs an additional 15-20 years before the patient descends into dementia.(38,39) The latter half of this trajectory is similar or slightly shorter in carriers of determinative mutations, based on dating to the age at which a carrier’s affected parent exhibited the earliest symptoms of incipient dementia and the use of evidence of plaque on fluorbetapir PET.(17-19) Between the first appearance of plaque and the diagnosis of dementia, the decline in CSF Aβ42 accelerates, CSF tau begins to rise, age-related atrophy of the hippocampus and other grey matter increases, and brain metabolic activity on 2-DG PET begins to decline.((17-19,38,39), and personal communication, Dr. Adam Fleisher).
The staging of the emergence of plaque and the decline of CSF Aβ prior to the first suggestions of tau pathology and accelerated neuronal loss is consistent with a range of prior evidence from human autopsies and animal models(2,3) (see Figure 4). The appearance of Aβ much as 30 years before the onset of dementia, and of tau later on in the process but yet while the patient remains cognitively within the normal range, is consistent with evidence implicating both Aβ(37) and tau pathology in “normal” age-related cognitive decline. Similarly, the early disturbance of brain metabolism is consistent with evidence of impaired cerebral metabolism in carriers of the ApoEε4 allele and maternal history of AD that precedes AD-related symptoms by decades,(24-27) and of impaired cerebral metabolism correlated to Aβ deposits on PiB-PET preceding cognitive decline in the general aging population.(36)

Figure 4. Time-Course of Alzheimer’s Biomarkers and Underlying Pathological Process. Reproduced from (16), with permission. ©2009 Nature Publications Group.
Moreover, there is extensive evidence that the relationship between Aβ and tau in age-related neurodegenerative disease extends beyond the long-supported “cascade” model in which the former initiates the hyperphosphorylation and aggregation of the latter: even without hyperphosphorylation, tau acts as a mediator of Aβ-induced cognitive deficits and synaptic abnormalities in animal and culture studies.(28-32) And while removal of Aβ through immunotherapy can prevent and even reverse the earliest stages of tau hyperphosphorylation, it is not sufficient to effect the clearance of mature neurofibrillary tangles nor to reverse aspects of cognitive decline related to tau((32-35), and see Aβ vaccine trial neuropathological data reviewed here, here, and here).
Go Early — and Go Deep
The move by scientists and regulators to begin administering Aβ immunotherapy not only early in clinical dementia, but during the preclinical phase of the disease, is an extremely important development. The new regulatory posture responds to the recognition that the volunteers in these trials are not merely “at high risk of Alzheimer’s,” but are fated to dementia (and to the many other diseases and disabilities of aging) unless rescued by rejuvenation biotechnology — a fate, it must be emphasized, shared by us all, as part of the degenerative aging process.
The neurodegeneration of aging involves multiple cellular and molecular lesions, of which Aβ appears to play a distinctive role in cogntive decline, whether directly or as an early driver of other neurodegenerative processes.(2,3,17-23,28-32) Its early removal can be expected to decelerate the emergence of other cellular and molecular lesions of aging, but it would not arrest them; accordingly, clearance of Aβ pathology alone is insufficient to fully prevent cognitive decline, even if that cogntitive decline is itself initiated by Aβ.(32-35) If Aβ vaccines are to be used as monotherapies, they should ideally be initially administered even earlier intervention than is planned in the CAP trials. The long period of CNS Aβ accumulation prior to the point at which plaques become manifest and other neuropathological and functional events accelerate(17-19,38,39), as well as the involvement of Aβ(37) and tau pathology in “normal” age-related cognitive decline argue that a monotherapeutic approach can only be expected to arrest the long march toward Alzheimer’s disease if initiated as much as 30 years before the anticipated onset of dementia.
Furthermore, even the complete prevention of Aβ accumulation would not abrogate other metabolic and traumatic causes of the neurofibrillary pathology and loss of neurons that are normally associated with AD, let alone the intra- and extracellular aggregates that are more characteristic of other age-related neurodegenerative diseases.In fact, Aβ plaque, neurofibrillary tangles, Lewy bodies, and less-specific aggregates (such as Hirano bodies, intermediate filament inclusions, or TAR-DNA binding protein 43 pathology) are actually co-present in AD and in “normal” brain aging, and the proteins themselves are often physically intertwined with one another.(40,41) Indeed, the differential diagnosis of these diseases based on neuropathology is ultimately as much a conceptual convenience as a genuinely pathognomonic exercise: autopsy reveals which aspect(s) of brain aging have proceeded at the fastest rate in a given patient’s brain, but it is the very essence of brain aging that all of us over the age of 40 or 50 are in the prodromal phase of all of them.
Therefore, regulatory reform must also make allowance for the clinical trials of combinations of agents that have not undergone prior Phase2/3 testing as monotherapies, where a rational case can be built based on the etiopathogenesis of a particular age-related disease, where animal data appears promising, and where no specific safety concern exists. In AD, for instance, there is a strong argument for early testing of combinations of agents targeting both Aβ and tau pathology, and insisting on the separate testing of these two kinds of agents is likely to severely hamper the demonstration of the efficacy of either. There are increasing calls for such early testing of combination therapies in AD(7) and other diseases of aging,(42) and happily, regulators are listening. Two major stakeholder meetings have brought together AD patient and caregiver advocates, researchers, and FDA regulators, to work toward the development of new clinical trial designs and regulatory reforms to advance previously-untested combination therapies for AD into clinical testing. And just last month, FDA’s Center for Drug Evaluation and Research (CDER) issued the latest draft text of “Guidance for Industry on Codevelopment of Two or More New Investigational Drugs for Use in Combination.
What is needed is an all-out push for a comprehensive suite of rejuvenation biotechnologies, to remove, replace, repair and render harmless the cellular and molecular damage of aging. As regenerative therapies against the diseases and disabilities of aging demonstrate their readiness for clinical trials, regulations must be tailored to as to allow studies to recruit participants before they have crossed the threshold of clinical disease, and in rational combinations based on the known aetiology and pathogenesis of specific clinical entities. These scientific and regulatory advances in AD research must now be used as precedents and roadmaps toward similar acommodations across the range of the diseases and disabilities of aging.
Science, advocacy, and regulatory reform have come together to save minds and lives in AD. The same strategies, reasoning, and action must now be brought to bear against all forms of age-related ill health, until all are under medical control and youth and health are our birthright.
References
1. Brookmeyer R, Johnson E, Ziegler-Graham K, Arrighi HM. Forecasting the global burden of Alzheimer’s disease. Alzheimers Dement. 2007 Jul;3(3):186-91. doi: 10.1016/j.jalz.2007.04.381. PubMed PMID: 19595937.
2. Selkoe DJ. Resolving controversies on the path to Alzheimer’s therapeutics. Nat Med. 2011 Sep 7;17(9):1060-5. doi: 10.1038/nm.2460. Erratum in: Nat Med. 2011 Dec;17(12):1693. Nat Med. 2011 Nov;17(11):1521. PubMed PMID: 21900936.
3. Holtzman DM, Morris JC, Goate AM. Alzheimer’s disease: the challenge of the second century. Sci Transl Med. 2011 Apr 6;3(77):77sr1. doi: 10.1126/scitranslmed.3002369. Review. PubMed PMID: 21471435; PubMed Central PMCID: PMC3130546.
4. DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2001 Jul 17;98(15):8850-5. Epub 2001 Jul 3. PubMed PMID: 11438712; PubMed Central PMCID: PMC37524.
5. Hake A, Siemers E, Carlson C, Estergard W, Sundell K, Henley D, Eads, J Sexton C, Liu-Seifert H, Sethuraman G, Chen Y-F, Dean R, Willis B, DeMattos R, Mohs R. Efficacy and Safety of Intravenous Solanezumab in Patients with Mild to Moderate Alzheimer’s Disease: Results of Two Phase 3 Studies. American Academy of Neurology’s 65th AAN Annual Meeting in San Diego. Neurology. 2013 Feb 12;80(Meeting Abstracts 1):S24.006 And see reporting here and here.
6. Doody RS. Safety and efficacy of solanezumab in patients with mild to moderate Alzheimer’s Disease: results of phase 3. 5th conference clinical trials on Alzheimer’s disease: October 29-31, 2012, Grimaldi Forum, Convention Center, Monte Carlo. J Nutr Health Aging. 2012;16(9):801-2. doi: 10.1007/s12603-012-0393-5. PubMed PMID: 23131822. And see reporting here and here, and especially (10) below.
7. Aisen P, Sperling R Symposium 5: Solanezumab Phase 3 Results Audio Recording. Clinical Trials for Alzheimer’s disease (CTaD) 2012 Media. Online Resource. Accessed 2012-11-07.
8. Vellas B, Carrillo MC, Sampaio C, Brashear HR, Siemers E, Hampel H, Schneider LS, Weiner M, Doody R, Khachaturian Z, Cedarbaum J, Grundman M, Broich K, Giacobini E, Dubois B, Sperling R, Wilcock GK, Fox N, Scheltens P, Touchon J, Hendrix S, Andrieu S, Aisen P; EU/US/CTAD Task Force Members. Designing drug trials for Alzheimer’s disease: What we have learned from the release of the phase III antibody trials: A report from the EU/US/CTAD Task Force. Alzheimers Dement. 2013 Jul;9(4):438-44. doi: 10.1016/j.jalz.2013.03.007. PubMed PMID: 23809364.
9. Siemers E. Solanezumab phase 3 results: implications for Alzheimer’s disease modification. 5th conference clinical trials on Alzheimer’s disease: October 29-31, 2012, Grimaldi Forum, Convention Center, Monte Carlo. J Nutr Health Aging. 2012;16(9):807-8. doi: 10.1007/s12603-012-0393-5. PubMed PMID: 23131822.
10. Strobel G. CTAD: New Data on Sola, Bapi, Spark Theragnostics Debate. Alzforum. 2012 Nov 9. Online Resource. Accessed 2013-07-19
11. Farlow M, Arnold SE, van Dyck CH, Aisen PS, Snider BJ, Porsteinsson AP, Friedrich S, Dean RA, Gonzales C, Sethuraman G, DeMattos RB, Mohs R, Paul SM, Siemers ER. Safety and biomarker effects of solanezumab in patients with Alzheimer’s disease. Alzheimers Dement. 2012 Jul;8(4):261-71. doi: 10.1016/j.jalz.2011.09.224. Epub 2012 Jun 5. PubMed PMID: 22672770.
12. Feldman H, Weninger S. Presymptomatic treatment trials: A moderated panel discussion. Alzheimers Dement. 2012 Jul;8(4 Suppl):P426-P427.
13. Bohrmann B, Baumann K, Benz J, Gerber F, Huber W, Knoflach F, Messer J, Oroszlan K, Rauchenberger R, Richter WF, Rothe C, Urban M, Bardroff M, Winter M, Nordstedt C, Loetscher H. Gantenerumab: a novel human anti-Aβ antibody demonstrates sustained cerebral amyloid-β binding and elicits cell-mediated removal of human amyloid-β. J Alzheimers Dis. 2012;28(1):49-69. doi: 10.3233/JAD-2011-110977. PubMed PMID: 21955818.
14. Ostrowitzki S, Deptula D, Thurfjell L, Barkhof F, Bohrmann B, Brooks DJ, Klunk WE, Ashford E, Yoo K, Xu ZX, Loetscher H, Santarelli L. Mechanism of amyloid removal in patients with Alzheimer disease treated with gantenerumab. Arch Neurol. 2012 Feb;69(2):198-207. doi: 10.1001/archneurol.2011.1538. Epub 2011 Oct 10. PubMed PMID: 21987394.
15. Citron M. Strategies for disease modification in Alzheimer’s disease. Nat Rev Neurosci. 2004 Sep;5(9):677-85. Review. PubMed PMID: 15322526.
16. Perrin RJ, Fagan AM, Holtzman DM. Multimodal techniques for diagnosis and prognosis of Alzheimer’s disease. Nature. 2009 Oct 15;461(7266):916-22. doi: 10.1038/nature08538. Review. PubMed PMID: 19829371; PubMed Central PMCID: PMC2810658.
17. Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, Marcus DS, Cairns NJ, Xie X, Blazey TM, Holtzman DM, Santacruz A, Buckles V, Oliver A, Moulder K, Aisen PS, Ghetti B, Klunk WE, McDade E, Martins RN, Masters CL, Mayeux R, Ringman JM, Rossor MN, Schofield PR, Sperling RA, Salloway S, Morris JC; Dominantly Inherited Alzheimer Network. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med. 2012 Aug 30;367(9):795-804. doi: 10.1056/NEJMoa1202753. Epub 2012 Jul 11. Erratum in: N Engl J Med. 2012 Aug 23;367(8):780. PubMed PMID: 22784036; PubMed Central PMCID: PMC3474597.
18. Reiman EM, Quiroz YT, Fleisher AS, Chen K, Velez-Pardo C, Jimenez-Del-Rio M, Fagan AM, Shah AR, Alvarez S, Arbelaez A, Giraldo M, Acosta-Baena N, Sperling RA, Dickerson B, Stern CE, Tirado V, Munoz C, Reiman RA, Huentelman MJ, Alexander GE, Langbaum JB, Kosik KS, Tariot PN, Lopera F. Brain imaging and fluid biomarker analysis in young adults at genetic risk for autosomal dominant Alzheimer’s disease in the presenilin 1 E280A kindred: a case-control study. Lancet Neurol. 2012 Dec;11(12):1048-56. doi: 10.1016/S1474-4422(12)70228-4. Epub 2012 Nov 6. PubMed PMID: 23137948.
19. Fleisher AS, Chen K, Quiroz YT, Jakimovich LJ, Gomez MG, Langois CM, Langbaum JB, Ayutyanont N, Roontiva A, Thiyyagura P, Lee W, Mo H, Lopez L, Moreno S, Acosta-Baena N, Giraldo M, Garcia G, Reiman RA, Huentelman MJ, Kosik KS, Tariot PN, Lopera F, Reiman EM. Florbetapir PET analysis of amyloid-β deposition in the presenilin 1 E280A autosomal dominant Alzheimer’s disease kindred: a cross-sectional study. Lancet Neurol. 2012 Dec;11(12):1057-65. doi: 10.1016/S1474-4422(12)70227-2. Epub 2012 Nov 6. PubMed PMID: 23137949; PubMed Central PMCID: PMC3515078.
20. Leverenz JB, Watson GS, Shofer J, Zabetian CP, Zhang J, Montine TJ. Cerebrospinal fluid biomarkers and cognitive performance in non-demented patients with Parkinson’s disease. Parkinsonism Relat Disord. 2011 Jan;17(1):61-4. doi: 10.1016/j.parkreldis.2010.10.003. Epub 2010 Nov 1. PubMed PMID: 21044858; PubMed Central PMCID: PMC3071582.
21. Kalaitzakis ME, Walls AJ, Pearce RK, Gentleman SM. Striatal Aβ peptide deposition mirrors dementia and differentiates DLB and PDD from other parkinsonian syndromes. Neurobiol Dis. 2011 Feb;41(2):377-84. doi: 10.1016/j.nbd.2010.10.005. Epub 2010 Oct 14. PubMed PMID: 20951207.
22. Alves G, Brønnick K, Aarsland D, Blennow K, Zetterberg H, Ballard C, Kurz MW, Andreasson U, Tysnes OB, Larsen JP, Mulugeta E. CSF amyloid-beta and tau proteins, and cognitive performance, in early and untreated Parkinson’s disease: the Norwegian ParkWest study. J Neurol Neurosurg Psychiatry. 2010 Oct;81(10):1080-6. doi: 10.1136/jnnp.2009.199950. Epub 2010 Jun 14. PubMed PMID: 20547614.
23. Kasuga K, Tokutake T, Ishikawa A, Uchiyama T, Tokuda T, Onodera O, Nishizawa M, Ikeuchi T. Differential levels of alpha-synuclein, beta-amyloid42 and tau in CSF between patients with dementia with Lewy bodies and Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2010 Jun;81(6):608-10. doi: 10.1136/jnnp.2009.197483. PubMed PMID: 20522869.
24. Langbaum JB, Chen K, Caselli RJ, Lee W, Reschke C, Bandy D, Alexander GE, Burns CM, Kaszniak AW, Reeder SA, Corneveaux JJ, Allen AN, Pruzin J, Huentelman MJ, Fleisher AS, Reiman EM. Hypometabolism in Alzheimer-affected brain regions in cognitively healthy Latino individuals carrying the apolipoprotein E epsilon4 allele. Arch Neurol. 2010 Apr;67(4):462-8. doi: 10.1001/archneurol.2010.30. PubMed PMID: 20385913; PubMed Central PMCID: PMC2943432.
25. Thambisetty M, Beason-Held L, An Y, Kraut MA, Resnick SM. APOE epsilon4 genotype and longitudinal changes in cerebral blood flow in normal aging. Arch Neurol. 2010 Jan;67(1):93-8. doi: 10.1001/archneurol.2009.913. PubMed PMID: 20065135; PubMed Central PMCID: PMC2856443.
26. Mosconi L, Mistur R, Switalski R, Brys M, Glodzik L, Rich K, Pirraglia E, Tsui W, De Santi S, de Leon MJ. Declining brain glucose metabolism in normal individuals with a maternal history of Alzheimer disease. Neurology. 2009 Feb 10;72(6):513-20. doi: 10.1212/01.wnl.0000333247.51383.43. Epub 2008 Nov 12. PubMed PMID: 19005175; PubMed Central PMCID: PMC2677512.
27. Reiman EM, Chen K, Alexander GE, Caselli RJ, Bandy D, Osborne D, Saunders AM, Hardy J. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer’s dementia. Proc Natl Acad Sci U S A. 2004 Jan 6;101(1):284-9. Epub 2003 Dec 19. PubMed PMID: 14688411; PubMed Central PMCID: PMC314177.
28. Roberson ED, Halabisky B, Yoo JW, Yao J, Chin J, Yan F, Wu T, Hamto P, Devidze N, Yu GQ, Palop JJ, Noebels JL, Mucke L. Amyloid-β/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J Neurosci. 2011 Jan 12;31(2):700-11. doi: 10.1523/JNEUROSCI.4152-10.2011. PubMed PMID: 21228179; PubMed Central PMCID: PMC3325794.
29. Vossel KA, Zhang K, Brodbeck J, Daub AC, Sharma P, Finkbeiner S, Cui B, Mucke L. Tau reduction prevents Aβ-induced defects in axonal transport. Science. 2010 Oct 8;330(6001):198. doi: 10.1126/science.1194653. Epub 2010 Sep 9. PubMed PMID: 20829454; PubMed Central PMCID: PMC3024010.
30. Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wölfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Götz J. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell. 2010 Aug 6;142(3):387-97. doi: 10.1016/j.cell.2010.06.036. Epub 2010 Jul 22. PubMed PMID: 20655099.
31. Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science. 2007 May 4;316(5825):750-4. PubMed PMID: 17478722.
32. Oddo S, Vasilevko V, Caccamo A, Kitazawa M, Cribbs DH, LaFerla FM. Reduction of soluble Aβ and tau, but not soluble Aβ alone, ameliorates cognitive decline in transgenic mice with plaques and tangles. J Biol Chem. 2006 Dec 22;281(51):39413-23. Epub 2006 Oct 20. PubMed PMID: 17056594.
33. Serrano-Pozo A, William CM, Ferrer I, Uro-Coste E, Delisle MB, Maurage CA, Hock C, Nitsch RM, Masliah E, Growdon JH, Frosch MP, Hyman BT. Beneficial effect of human anti-amyloid-beta active immunization on neurite morphology and tau pathology. Brain. 2010 May;133(Pt 5):1312-27. doi: 10.1093/brain/awq056. Epub 2010 Mar 31. PubMed PMID: 20360050; PubMed Central PMCID: PMC2859150.
34. Nicoll JA, Barton E, Boche D, Neal JW, Ferrer I, Thompson P, Vlachouli C, Wilkinson D, Bayer A, Games D, Seubert P, Schenk D, Holmes C. Aβ species removal after Aβ42 immunization. J Neuropathol Exp Neurol. 2006 Nov;65(11):1040-8. PubMed PMID: 17086100.
35. Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM. Aβ immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron. 2004 Aug 5;43(3):321-32. PubMed PMID: 15294141.
36. Clark VH, Resnick SM, Doshi J, Beason-Held LL, Zhou Y, Ferrucci L, Wong DF, Kraut MA, Davatzikos C. Longitudinal imaging pattern analysis (SPARE-CD index) detects early structural and functional changes before cognitive decline in healthy older adults. Neurobiol Aging. 2012 Dec;33(12):2733-45. doi: 10.1016/j.neurobiolaging.2012.01.010. Epub 2012 Feb 24. PubMed PMID: 22365049.
37. Doré V, Villemagne VL, Bourgeat P, Fripp J, Acosta O, Chetélat G, Zhou L, Martins R, Ellis KA, Masters CL, Ames D, Salvado O, Rowe CC. Cross-sectional and Longitudinal Analysis of the Relationship Between Aβ Deposition, Cortical Thickness, and Memory in Cognitively Unimpaired Individuals and in Alzheimer Disease. JAMA Neurol. 2013 Jul 1;70(7):903-11. doi: 10.1001/jamaneurol.2013.1062. PubMed PMID: 23712469.
38. Jack CR Jr, Wiste HJ, Lesnick TG, Weigand SD, Knopman DS, Vemuri P, Pankratz VS, Senjem ML, Gunter JL, Mielke MM, Lowe VJ, Boeve BF, Petersen RC. Brain β-amyloid load approaches a plateau. Neurology. 2013 Mar 5;80(10):890-6. doi: 10.1212/WNL.0b013e3182840bbe. Epub 2013 Feb 27. PubMed PMID: 23446680; PubMed Central PMCID: PMC3653215.
39. Villemagne VL, Burnham S, Bourgeat P, Brown B, Ellis KA, Salvado O, Szoeke C, Macaulay SL, Martins R, Maruff P, Ames D, Rowe CC, Masters CL; Australian Imaging Biomarkers and Lifestyle (AIBL) Research Group. Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: a prospective cohort study. Lancet Neurol. 2013 Apr;12(4):357-67. doi: 10.1016/S1474-4422(13)70044-9. Epub 2013 Mar 8. PubMed PMID: 23477989.
40. Armstrong RA, Lantos PL, Cairns NJ. What determines the molecular composition of abnormal protein aggregates in neurodegenerative disease? Neuropathology. 2008 Aug;28(4):351-65. doi: 10.1111/j.1440-1789.2008.00916.x. Epub 2008 Apr 23. Review. PubMed PMID: 18433435.
41. Armstrong RA, Lantos PL, Cairns NJ. Overlap between neurodegenerative disorders. Neuropathology. 2005 Jun;25(2):111-24. Review. PubMed PMID: 15875904.
42. de Grey AD. The future dominance of combination therapies: implications for today’s medical research. Rejuvenation Res. 2012 Oct;15(5):443-4. doi: 10.1089/rej.2012.1376. PubMed PMID: 22989287.
43. de Grey AD. Has Hippocrates had his day? Rejuvenation Res. 2006 Fall;9(3):371-3. PubMed PMID: 16859477.
44. Joshi AD, Pontecorvo MJ, Clark CM, Carpenter AP, Jennings DL, Sadowsky CH, Adler LP, Kovnat KD, Seibyl JP, Arora A, Saha K, Burns JD, Lowrey MJ, Mintun MA, Skovronsky DM; Florbetapir F 18 Study Investigators. Performance characteristics of amyloid PET with florbetapir F 18 in patients with alzheimer’s disease and cognitively normal subjects. J Nucl Med. 2012 Mar;53(3):378-84. doi: 10.2967/jnumed.111.090340. Epub 2012 Feb 13. PubMed PMID: 22331215.