
The aging brain is characterized by the accumulation of a variety of proteinaceous aggregates, high levels of which constitute the distinctive neuropathological hallmarks and (in the consensus view) the underlying drivers of the neurodegenerative diseases of aging. These include β-amyloid (Aβ) in Alzheimer’s disease (AD); α-synuclein (AS) aggregates in Parkinson’s disease (PD), Lewy body dementia, and other synucleinopathies; and neurofibrillary tangles in AD and frontotemporal dementia (FTD). Even prior to the eruption of such severe and diagnosable clinical conditions, however, Aβ[1,52-58] and aggregated α-synuclein and tau are implicated in driving “normal” age-related cognitive decline.
Removal of these aggregates from the brain is therefore a central damage-repair strategy to prevent and arrest the course of “normal” cognitive aging and its diagnostically-specified extreme manifestations. Happily, this subfield of rejuvenation research has been advanced further toward medical availability than any other, with new strategically-positioned trials of Aβ immunotherapies and of a first-in-class α-synuclein vaccine. Other rejuvenation biotechnologies that will be key to maintaining healthy cognitive function and averting neurodegenerative disease are also advancing along the development pipeline — notably, immunotherapeutic approaches to the removal of pathological tau species (please see several previous blog posts on the subject for examples).
Another pillar of comprehensive neurorejuvenation is cell therapy for the aging brain. Cell therapy using mixed fetal ventral mesencephalic cell populations was administered to humans with PD in small, preliminary trials thirty years ago, with inconsistent but promising effect.[3] After a long hiatus, this approach is being revived in the recently-initiated TRANSEURO trial. Along with progress in applying induced pluripotent stem cell (iPS) technology in nonhuman primate models of the disease[2,3] and improved transplantation protocols that bring us closer to functional restoration of dopaminergic circuitry,[4] there is strong grounds for optimism that repair of the sine qua non degenerative lesion in PD will soon be achieved.

Change in 18F-Labeled Dopamine Uptake in the Brains of PD Patients After Embryonic Dopaminergic Neuron Transplantation. Reproduced from [36] with permission. © 2001 Massachusetts Medical Society.
However, cell therapy to preserve and restore the neuronal circuitry underlying higher-order cognitive functions in the aging brain is a much more formidable undertaking. Unlike with other major organs such as the heart or kidneys — or even the repair of dopaminergic brain circuitry as exhibited prominently in PD — wholesale replacement of brain functional units is undesirable, due to the structural basis of memory and identity. Thus, a more sophisticated and gradualist approach is required, in which existing circuits are rebuilt and ongoingly reinforced by ongoing integration of transplanted neurons and precursors, in such a way as to maintaining their existing architecture. Such an approach is indeed used by the brain in neurogenesis, but most evidence suggests that endogenous neurogenesis is confined to a limited number of brain regions.[6] Furthermore, most of the neurons generated through neurogenesis are transient and non-integrating,[6] and the rate of neuronal replacement falls precipitously in the postnatal period in humans (particularly as compared to mice).[11,12] The neurogenic/neuroregenerative functions of resident stem and progenitor cell populations depend on the specialized microenvironments of their niches and of the “soil” of their remote migration sites, and areas of the brain that are not replenished by neurogenic pathways appear to lack the conditions to integrate transplanted cells.[6,9] Critically, these non-neurogenic areas include the neocortex, cerebellum, and the basal forebrain, where AD tau pathology first arises and where the loss cholinergic neurons closely parallels cognitive decline.[6] Thus, successful cell replacement requires strategies that do not depend on simply exploiting and augmenting existing neurogenic processes for the repair and prevention of age-related cognitive degeneration.
The brain’s mobilization of stem and progenitor cells in response to more severe, acute insults is also an unpromising endogenous model for approaches to brain rejuvenation. Traumatic brain injury and stroke elicit an acute burst in neurogenic activity in neurogenic areas of the brain, followed by in-migration of newborn neurons and progenitors into the site of injury (ischaemic striatum, in the case of most stroke models). However, very few of these newly-generated neurons survive, and there is yet no convincing evidence that they integrate into damaged neuronal circuitry and participate in structural repair. Instead, their primary role appears to be limited to providing transient trophic support, to limit the attrition of neurons that survive the initial insult.[6,7,10]
If the neurogenic explosion that follows an acute insult is of disappointingly little reconstructive effect, there is thus far no evidence at all of an endogenous neurogenic response to the gradual age-related attrition of neurons. For example, selective neuronal apoptosis of neocortex neurons induced as a model of such attrition leads to no such response.[5] More promisingly, stem and progenitor cells that are transplanted into animal models of stroke do seem to participate in circuit repair and enhance functional restoration.[6,7] But the same cannot be said in animal models of AD, in which neuronal stem and progenitor cells that are transplanted into the basal forebrain do not appear to contribute directly to repair of local circuitry, though they do provide trophic support for surviving native neurons.[6,8]
While strategies to overcome these limits to native regenerative capacity are under pursuit,[6-9] the use of cell therapy to maintain and rebuild aging and AD brain circuitry clearly constitutes one of the greatest and longest-term challenges to comprehensive human rejuvenation.
Thus, a therapeutic chasm appears to be opening between the pace of development of two key rejuvenation biotechnologies that will be required to maintain youthful cognitive function and prevent age-related neurodegenerative disease. On the one hand, rejuvenation biotechnologies targeting the removal of the main proteinaceous aggregates are being pursued by multiple groups, and several are already in late-stage human clinical testing. On the other hand, very little obvious progress has been made in cell replacement therapy in the brain, particularly if we confine ourselves to consideration of that fraction of such work that is aimed at the intrinsically challenging work of maintaining and rebuilding neuronal circuitry in areas involved in higher cognitive function. No such work is in clinical testing, or indeed being explored in nonhuman primates, and only very limited work has been pursued in mice.
Thus, there exists a risk that a “doughnut hole” may emerge in the prevention and treatment of dementias of aging, in which aggregate-clearing therapies may soon be available for individuals at high risk of dementia (and not long thereafter for the wider aging population, granted political pressure and ongoing regulatory reform), but in which the respite granted by these therapies is short-lived due to a mixture of pre-existing and ongoing (if slowed) neuronal loss, due to the lack of mature neuronal replacement and reinforcement biotechnologies. What might be done to help patients leap across this gap in the therapeutic bridge?
A Bridging Strategy: Neuroprotective Agents
Neuroprotective agents offer a potential stopgap, to hold the therapeutic window open in the period between the availability of aggregate-clearing immunotherapies and the development of neuronal replacement techniques. From first principles, one might anticipate that in a scenario in which therapeutic clearance of some aggregate had been achieved, the most effective neuroprotective agents would be those that target mechanisms of neurodegeneration that are not directly downstream of these aggregates. However, it is also likely that in the earliest iterations of these therapies, their specificity, range of action, therapeutic index, pharmacokinetics, or other properties may limit the scope and magnitude of clearance that can be achieved in a given round of application, so that even agents that allow vulnerable neurons to survive the downstream effects of these aggregates may yet deliver some neuroprotective benefit.
Heretofore, the most commonly pursued targets for neuroprotective agents have been oxidative stress, inflammation, and excitotoxicity. There has already been a significant amount of research on agents that modulate these pathways in animal models, and in some cases even research in humans.[13] Numerous existing drugs in clinical use and dietary supplements modulate these pathways, and researchers have probed their potential use as AD or dementia prophylactics through epidemiological research and secondary analyses of clinical trials originally conducted for drugs’ approved indications. In several cases, such research had supported the potential neuroprotective effects of existing agents in reducing the risk or progression of cognitive decline, AD, or total dementia (reviewed in [13]). To date, however, every time that one of those agents that has been subjected to clinical trials have failed to show an effect on with AD or other age-related cognitive decline. These include simvastatin and atorvastin (HMG CoA-reductase inhibitors), Ginko biloba (an herbal extract), several non-steroidal anti-inflammatory drugs, rosiglitazone (antidiabetic (agonist of peroxisome proliferator-activated receptor γ)), α-tocopherol (the most common supplemental form of vitamin E (antioxidant)), selenium (antioxidant, apoptosis modulator), and docosahexanoic acid (DHA) (omega-3 fatty acid in fatty fish) (see references in [13]).
Image provided by NIH Common Fund.
Additionally, memantine was for a time anticipated to protect against excitotoxic cell death in AD and other neurodegenerative disorders. Memantine, an approved for treatment for AD that is mildly effective for symptomatic treatment of moderate to late-stage disease, is an antagonist of the glutaminergic NMDA receptor. Neurons are thought to be vulnerable to inappropriate activation of calcium channels regulated by these receptors in AD, PD, and in the post-stroke and -TBI condition; possible reasons for this include the inability of affected neurons to maintain normal ionic polarization because mitochondrial dysfunction or high energetic demands, or direct interactions of Aβ with NMDA receptors. These effects may impede the effects of the receptor’s physiological blocker, Mg2+, lowering the threshold level of glutamate required for Ca2+ influx, resulting in cellular toxicity and death.[14,15] In cell models, memantine exhibits a rapid off-rate constant with the NMDA receptor and relatively strong voltage dependency of action, allowing it to block the receptor under pathological hyperactivation yet remain dissociated during periods of physiological activation.[14,15] Consistent with this, memantine exhibited neuroprotective effects against excitotoxic insults in animal models.[14,15] Unfortunately, no evidence of a neuroprotective, disease-modifying effect of memantine has emerged in clinical trials or observational studies in humans.[15,30]
A parallel case in PD is the dopamine D2/D3 receptor agonist pramipexole (Mirapex®), a drug already approved for use in PD. Like memantine, pramipexole exhibited neuroprotective effects in cell culture and animal studies models of dopaminergic neurotoxicity. Yet people with PD receiving pramipexole or placebo exhibited similar rates of decline in clinical scores and of striatal dopamine-transporter binding assessed on SPECT over the course of a 15-month placebo-controlled trial.[21]
Nonetheless, researchers continue to pursue potential approaches for neuroprotective therapeutics, targeting these well-trodden pathways as well as others emergent, such as brain-derived neurotrophic factor (BDNF)[16] and other neurotrophins; transforming growth factor-β1 (TGF-β1);[17] novel neuroimmodulatory strategies such as targeting the chemokine CCL5/RANTES;[18] the galanin pathway;[19] the nuclear enzyme poly(ADP-ribose) polymerase 1 (PARP-1);[20] and (in PD) agonism of glucagon-like peptide-1.[50]
A New Target PERKs UP(R)
Human autopsy studies and experimental work have revealed that neuronal endoplasmic reticulum (ER) stress is increased early in neurodegenerative diseases of aging such as AD,[22-24,66] PD,[23,25,26] and genetic- and age-related tauopathies (diseases of accumulation of aberrant tau species).[24,27,66] (and see [28] for a recent review). Amongst other functions, the ER is a center for the folding, assembly, and quality control of secretory, organellar, and transmembrane proteins. When its protein-folding machinery is overwhelmed, un- or misfolded proteins begin to accumulate in the ER lumen, leading to ER stress. In response, the ER activates the unfolded protein response (UPR), which (a) induces a general translational block to reduce the production of proteins that would further tax its protein-folding capacity, (b) selectively increases the transcription of genes involved in its protein-folding and quality control machinery, and (c) accelerates the degradation of misfolded proteins (via the lysosome, and via the proteasome in a process known as ER-associated degradation (ERAD)).* While the UPR therefore can serve neuroprotective functions by resolving ER stress, elements of the UPR machinery can alternatively trigger apoptosis when the UPR proves insufficient to this task.[28,49]
Protein kinase RNA-like endoplasmic reticulum kinase (PERK) is one of three ER transmembrane proteins involved in detecting ER stress and regulating the UPR. Under conditions of ER stress, PERK phosphorylates the eukaryotic translation initiation factor 2α (eIF2α); phosphorylation of eIF2α leads to general translational arrest, while simultaneously selectively increasing the translation of activation of transcription-4 (ATF4), which in turn upregulates expression of UPR genes involved in antioxidant function, amino acid metabolism, autophagy, and apoptosis.[28] As discussed in more detail below, because several other cellular stressors also elicit an eIF2α-mediated cellular stress response, the UPR has been incorporated by some into a wider “Integrated Stress Response” (ISR).
Several studies have reported that as in age-related neurodegenerative disease, patients and animals with prion diseases exhibit engagement of UPR/ISR (reviewed in [28,31]). One model of prion disease is infection with murine Rocky Mountain Laboratory prion, an aggregating species that induces a transmissible spongiform encephalopathy. Mice with a transgenic threefold overexpression of the native murine prion protein (PrP) exhibit reduced hippocampal synaptic transmission and prion neuropathology 8 weeks after inocculation with RML, begin to show behavioral abnormalities 9 weeks, and die at the 12 week point (references in [31]). In 2012, researchers at the Universities of Leicester and of Nottingham, along with the MRC Laboratory of Molecular Biology in Cambridge, reported that levels of phosphorylated PERK (PERK-P) and eIF2α (eIF2α-P) in infected mouse hippocampus rose progressively with rising levels of total PrP and of the aggregated PrPSc conformer during the course of infection.[31]
UPR induction was an early event in the disease process: eIF2α-P levels began rising at 7 weeks post-infection, at which point the animals were asymptomatic, and several synaptic marker proteins were yet at normal levels. Despite this, synapse number had fallen to ~55% of control levels; the investigators suggest that this may have reflected impaired synaptic structural plasticity. At the 9-week mark, eIF2α-P levels rose markedly, even as levels of several synaptic marker proteins abruptly fell by ~50%, associated with a further reduction in synapse number. These structural effects were associated with clear functional impacts: for the first time, synaptic transmission declined, and behavioral abnormalities and spongiform neuropathology emerged (Figure 1). Consistent with the role of eIF2α-P in the UPR and with the abrupt reduction of synaptic proteins, general protein translation rates as measured by 35S-methionine incorporation into proteins in hippocampal slices were reduced by 50% in prion-infected vs. control mice. This was confirmed by reductions in the number of actively translating ribosomes and by Northern blots for selected mRNAs in individual polysomal fractions. Yet the specific translation of ATF4 mRNA was increased, again consistent with the elevation of eIF2α–P.[31] Only after this did neurodegeneration per se set in, with hippocampal pyramidal neuron counts suddenly declining by 50% at the 10-week mark (Figure 1). Animals died or were euthanized at 12 weeks.[31]
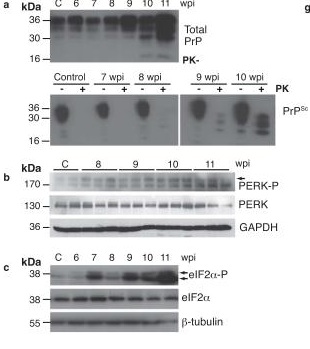

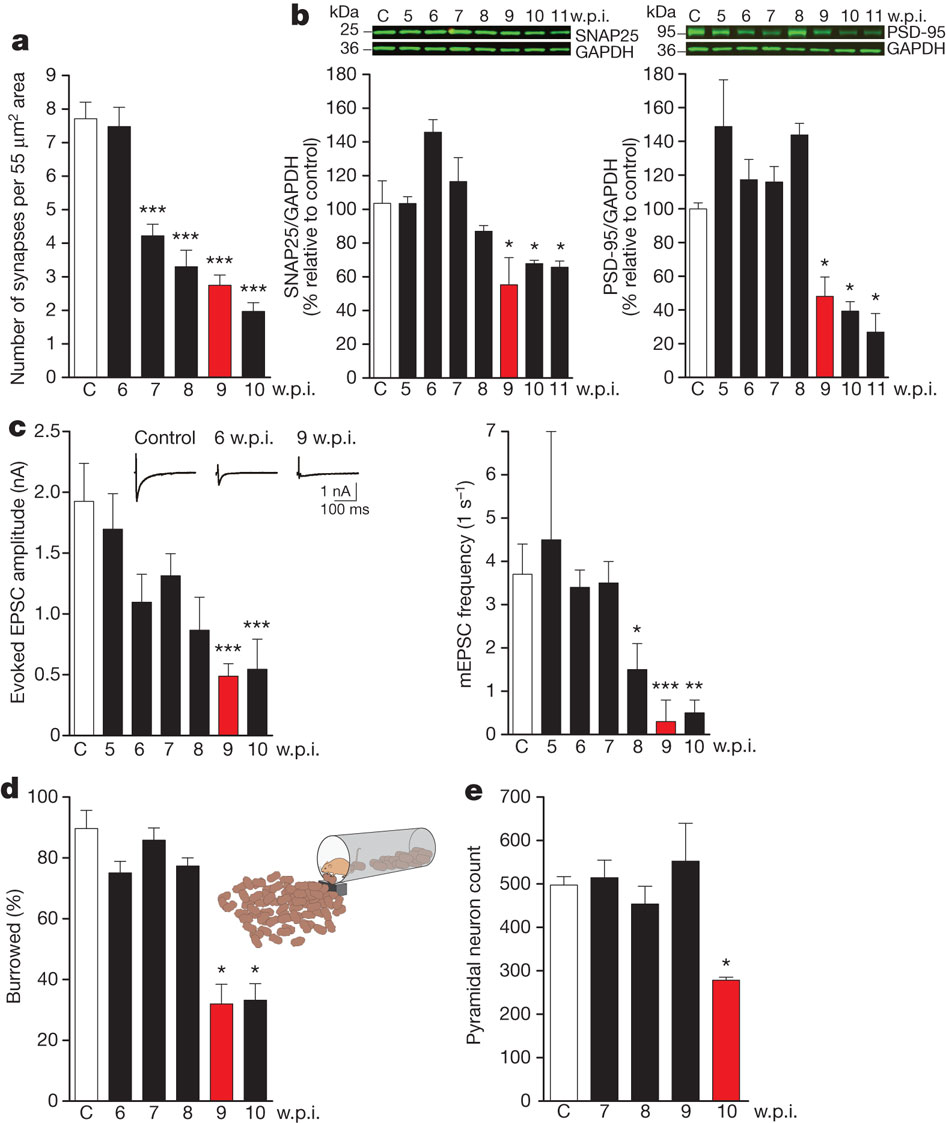
Figure 1. Induction of UPR via PERK-P and eIF2α-P Drives Neuronal Dysfunction and Loss in Prion-Infected Mice. Rising levels of PrP and PrpSc (first (a) above) is linked to appearance of phosphorylated PERK and eIF2α (first (b)) and general translational arrest (first (h,i)); this closely associates with synaptic loss (second (a)), fall in synaptic proteins (second (b)), synaptic dysfunction (c), and behavioral abnormality (d), and precedes neuronal loss (e). Reproduced from [31], with permission. © 2012 Nature Publications Group.
These findings suggested that some effect of the PERK-eIF2α arm of the UPR might be driving neuronal loss in murine prion disease. Indeed, the temporal association of eIF2α-P induction followed by mortality from prion disease seemed to be progressively advanced with increasing “doses” of available PrP. Thus, wild-type C57Bl6 mice exhibited elevated eIF2α-P at 16 weeks after RML inoculation, and died at ~22 weeks; eIF2α-P was induced at 9 weeks post-infection in hemizygous transgenics (with a 3-fold overexpression of PrP), and animals died at 9 weeks; and in homozygous transgenics, with six-fold overexpression of PrP, eIF2α-P was induced at 6 weeks post-infection, and mortality occurred at the 9-week mark.[31]
One finding did initially seem to undermine this hypothesis: the lack of evidence of apoptosis, increased autophagy, or even necrosis in hippocampal slices from infected mice, despite the fact that the eIF2α-P target gene ATF4 (which upregulates expression of genes involved in apoptosis and autophagy (the latter being central to the contested[32,33] phenomenon of “autophagic cell death”)).[31] Looking for an alternative explanation, the investigators noted the close association of general translational arrest with synaptic dysfunction, and the later emergence of neuronal death, and suggested that the UPR-induced translational arrest might itself be responsible for neurodegeneration in this setting.
To confirm the involvement of eIF2α-P and translational arrest in PrPSc-driven neuronal death, the investigators injected the hippocampi of prion-infected mice with lentiviral transgenes (LV) expressing either the specific eIF2α–P phosphatase GADD34, or short hairpin PrP (shPrP), or yellow fluorescent protein (YFP) as control. As would be expected, lentiviral GADD34 reduced eIF2α–P levels in infective animals relative to infected animals receiving no treatment or LV-YFP, without affecting levels of its upstream regulator PERK-P or of PrP. Lentiviral PrP reduced PrP levels and abrogated the rise in both PERK-P and eIF2α–P.[31] Crucially, reversing eIF2α phosphorylation with GADD34 was as effective knocking down PrP in preserving mRNA translation; synaptic number; and synaptic protein levels and transmission, and in ameliorating behavioral deficits. Additionally, of these treatments nearly abrogated neuronal loss and modestly increased survival.[31]
From Target to Treatment
Having identified the PERK-eIF2α translational arrest pathway as critical to neurodegeneration and behavioral abnormalities in prion-infected mice, the investigators went on to test whether pharmacological intervention in this pathway could exert similar neuroprotective effects. GSK2606414 is a highly specific inhibitor PERK’s phosphorylation of eIF2α, with good oral bioavailability and blood-brain barrier (BBB) penetrance in mice;[34] it was originally developed by the pharmaceutical company GSK to target PERK’s role in some cancers.[35]
Using mice with threefold overexpression of WT murine PrP and infected with RML, the investigators tested the ability of GSK2606414 to exert neuroprotective effects similar to those of lentiviral GADD34. Ambitiously, they tested its effects late in the course of the disease (9 weeks post-infection) as well as relatively early (7 weeks post-infection). As in their previous experiments,[31] the early-treatment group were already suffering synaptic loss at onset of treatment, but had not yet begun to exhibit neuronal loss or deficits in memory or behavior. By contrast, the late-onset treatment group were already suffering spongiform neuropathology by the time treatment began, and were exhibiting nonspecific behavioral and memory deficits.[34]
Several lines of evidence supported GSK2606414’s ability to inhibit activation of the PERK/eIF2α arm of the UPR in these animals. Hippocampal lysates from treated animals exhibited lower levels of phosphorylated PERK and eIF2α. The marked depression of protein synthesis rates that were observed in hippocampal slices of vehicle-treated animals were restored to the levels in uninfected controls, including both total synthesis rates (based on incorporation of 35S-methionine into protein) and the rates of production of key pre- and postsynaptic proteins (SNAP25 and PSD95, respectively). Yet translation of eIF2α targets ATF4 and the transcription factor C/EBP homologous protein (CHOP) (a key mediator of UPR-driven apoptosis) were selectively normalized. Contrariwise, there was no evidence of suppression of the other two arms of the UPR.[34]
By 12 weeks, all vehicle-treated control animals were exhibiting clinical signs specific to prion disease, including ataxia, sustained hunched posture, and hindlimb paralysis. At this point, by contrast, only 5/20 animals receiving early GSK2606414 treatment and 5/9 late-treated animals were exhibiting even nonspecific behavioral deficits (e.g. intermittent tail rigidity and hunching, or mild loss of coordination), and none were exhibiting definitive diagnostic indicators of prion disease: all treated animals continued to have normal posture, grooming, tail attitude, and hindlimb mobility.[34] Indeed, some of the nonspecific behavioral deficits that were observed in the late-treated animals were already present at onset of therapy, so that their persistence at 12 weeks shows only that the drug was unable to reverse existing nonspecific deficits.[34]
Similarly, at 9 weeks after RML inoculation, vehicle-treated and late-treated animals suffered a loss of object recognition memory and exhibited deficits in burrowing behavior. Late-onset GSK2606414 treatment did not restore lost object recognition, but early treatment completely prevented it; moreover, not only did treatment with GSK2606414 prevented burrowing behavioral deficits when administered early, but reversed those that had appeared at treatment onset in the late-treated group.[34]
GSK2606414 also afforded neuroprotective effects at multiple levels. GSK2606414-treated animals were almost completely shielded from prion-disease-driven neuronal loss, particularly in the hippocampus: vehicle-treated animals sustained a ~70% loss of CA1 pyramidal neurons at 12 weeks post-infection, whereas both early- and late-treated animals preserved as many neurons as uninfected, healthy control animals (see Figure 2).[34] Early treatment also almost completely prevented PrP-driven spongiosis across multiple brain regions, and while protection was incomplete in late-treated animals, the latter group suffered substantially less spongiosis than vehicle-fed controls, despite having already undergone some spongiform degeneration upon initiation of treatment (see Figure 2). Inflammatory activation of astrocytes was also greatly diminished in all treated animals, as measured by glial fibrillary acidic protein (GFAP) immunostaining.[34]
Extension to Models of Age-Related Neurodegeneration
Moreno et al‘s results[34] are promising for prion disease, of course. And because there is extensive evidence of ER stress and aberrant, sustained activation of the UPR in the neurological diseases of aging (such as AD, PD, and non-AD tauopathies[22-28]), they also suggest the possible use of this approach as a stopgap measure in the interregnum between the availability of immunotherapeutic clearance of the neuropathology driving these diseases and the advent of mature neuronal replacement therapy. More direct support is offered by several less-heralded but roughly contemporaneous reports that evaluated the effects of genetic or pharmacological inhibition of the PERK/eIF2α arm of the UPR in animal models of age-related brain disorders. Individually and together, these various investigations build the case for UPR inhibition as a broad-based neuroprotective strategy in the neurodegenerative aging process.
AD Mice
Writing in Nature Neuroscience, researchers led by Tao Ma in Eric Klann’s laboratory at New York University report that genetic inhibition of eIF2α phosphorylation substantially reduces the neurotoxic effects of Aβ in vitro, and in vivo in both otherwise-wild-type and in transgenic AD model mice.[37] Consistent with previous reports, the investigators found that the suppressive effect of Aβ oligomers on long-term potentiation (LTP) in hippocampal slices from wild-type mice is associated with inhibition of new neuronal protein synthesis and with sustained elevations in eIF2α-P. But selective postnatal knockout of PERK in hippocampal and forebrain neurons abrogated these abnormalities. Completing the evidential cycle, the rescue afforded by PERK knockout was itself prevented when hippocampal slices were treated with a pharmacological inhibitor of eIF2α dephosphorylation prior to exposure to Aβ and LTP-stimulating frequencies.[37]
Again consistent with earlier findings, Ma et al found evidence of sustained activation of the UPR in human AD autopsy brain and in AD model mice. As compared to WT mice, their 10–12-month-old transgenic APPswe/PS1ΔE9 mice exhibited an ~150% increase of eIF2α-P in the hippocampus and prefrontal cortex, and a similar elevation of ATF4.[37] Additionally, overall protein production was reduced, consistent with generalized inhibition of translation. But when these AD model mice were crossed with conditional PERK knockout mice, the PERK knockout mouse hippocampi exhibited eIF2α-P and ATF4 levels that were not only lower than those in control AD mouse brain, but lower than in fully WT animals. Similarly, knockout of PERK in AD mice increased protein synthesis rates to levels equivalent to or greater than those in WT brain (although the elevation in CHOP in these animals’ neurons persisted).[37] Again observations in previous reports in both human AD brains and the brains of AD model mice, APPswe/PS1ΔE9 mice exhibited abnormally elevated levels of postsynaptic density protein (PSD95) and depressed levels of synaptophysin — aberrations that mark synaptic pathology. Broadly parallelling the effects of GSK2606414 in prion-infected mice,[34] PERK knockout restored these synaptic proteins to the levels of WT mice.[37]†
Remarkably, PERK knockout in model AD mice completely normalized cognitive performance on the Morris water maze (MWM) platform escape latency (Figure 2 (a)), MWM platform-free probe test (Figure2 (b) and (c)), object location (Figure 2 (e)), and Y water maze (YWM) platform choice (Figure 2 (f)). Notably, PERK knockout did not affect performance on a visible platorm test (Figure X(d)), where motivation, swimming, ability, or simple visual acuity play a greater role in successful task completion.[37] Conditional PERK knockout also normalized in vivo LTP. Supporting the importance of eIF2α phosphorylation in these effects, most of the benefits of selective postnatal PERK knockout were recapitulated in AD model mice with global global knockout of GCN2, an alternative eIF2α kinase active during amino acid deprivation.[37]

Figure 2. Postnatal PERK Knockout Rescues Cognitive Performance in AD Model Mice. Reproduced from [37], via Klann Laboratory website.
Invertebrate Models of Amyotrophic lateral sclerosis (ALS)
While not a disease of aging per se, ALS is a fatal neurodegenerative disease associated in most patients with accumulations of aggregated TAR DNA-binding protein 43 (TDP-43) in motor neurons; such aggregates are also present in affected neurons of patients with the dementing disorder frontotemporal lobar degeneration (FTLD) and less frequently in other age-related neurological disorders.[38] It was recently reported that TDP-43 overexpression in the yeast Saccharomyces cerevisiae and the fruitfly Drosophila melanogaster was associated with increased eIF2α-P; treatment with GSK2606414 reduced the neurotoxicity of TDP-43 overexpression in Drosophila and in primary rat cortical neurons.[39]
Enhancing Normal Memory?
Beyond its role in the sustained ER stress of neurodegenerative disease, eIF2α phosphorylation has previously been reported to play a role in inhibition of LTP and of long-term memory consolidation under normal conditions.[41-43] In addition to its generalized effect on protein translation, eIF2α-P has a more specific effect on LTP that is mediated via ATF4, which blocks transcription of genes driven by cAMP responsive element (CRE) promoters, which include many key regulators of synaptic plasticity and LTP.[51] Accordingly, genetic enhancement of PERK-dependent eIF2α phosphorylation in in hippocampal CA1 pyramidal cells inhibits CREB-dependent memory pathways and impairs LTP,[43] while mice heterozygous for defective eIF2α phosophorylation sites enjoy enhanced memory, and the threshold for eliciting long-term LTP in hippocampal slices from such mice is reduced.[41]
In order to probe the effects of selectively inhibiting eIF2α phosphorylation on neuronal survival and memory, researchers at the University of California at San Francisco, McGill University in Montréal, and Genentech designed a novel two-stage cell-based screening system. The first screen is used to identify inhibitors of PERK signaling, and then additional screens are used to negatively select compounds that induce ATF4 or that also inhibit the two alternative arms of the UPR.[44]
Twenty-eight candidates emerged from this screen out of a library of 106,281 compounds, and one such candidate possessed an exceptionally low IC50. Subsequent testing revealed that this candidate inhibits PERK signaling through ATF4 and uniquely sustains global protein translation during ER stress, despite increasing PERK activation. In fact, the drug’s mechanism of action was found to be downstream not only of PERK phosphorylation, but of all eIF2α kinases (including GCN2, haeme-regulated inhibitor kinase (HRI, induced by haeme insufficiency or heavy metals), and protein kinase R (PKR, induced by the presence of double-stranded RNA and thus viral infection). All of these alternative kinases act by phosphorylating eIF2α on a common serine, leading to the generalized arrest of protein synthesis and selective induction of ATF4, CHOP, and their downstream regulatory targets that results from phosphorylation by PERK as part of the UPR. Thus eIF2α acts as a central mediator of multiple cellular stress pathways, termed the “Integrated Stress Response” (ISR) (Figure 3). Because the action of the drug occurs downstream of eIF2α phosphorylation, the authors dubbed the inhibitor of eIF2α signaling ISRIB, for “Integrated Stress Response InhiBitor.”[44]
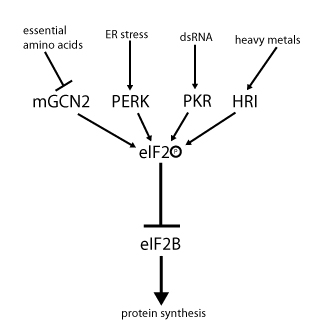
Figure 3. eIF2α at the Nexus of the Integrated Stress Response. Figure by Hannes Röst; reproduced under Creative Commons License from the Wikimedia Foundation.
{kind=link}
ISRIB was found to have good plasma pharmacokinetics after i.p. injection (Figure 4 A), excellent BBB penetrance (Figure 4 B), and to have a long (8 h) half-life. To test its effect on cognition in normal mice, Sidrauski et al treated animals with ISRIB or vehicle via i.p. injection, and then subjected all animals to a weak training regimen in the Morris water maze. After 5 days of training, ISRIB-treated mice were able to reach a hidden platform in just 16.4 ± 4.8 s, versus 68.1 ± 20 s in vehicle-treated animals (p<0.05) (Figure 4 C), and more strongly favored a learned but removed platform location in the probe test (Figure 4 D and E ). ISRIB treatment also enhanced long-term contextual ((Figure 4 F) and both short- and long-term auditory ((Figure 4 G) fear conditioning (assessed as percent of the test period spent immobile after the conditioned fear stimulus).

Figure 4. Effects of ISRIB on Measures of Cognition in Normal Mice. Plasma pharmacokinetics after i.p. injection (A); BBB penetrance (B); Morris water maze escape latency (C); probe test of quadrant recall (D, E); contextual (F) and auditory (G) fear conditioning (“CS” = “conditioned [fear] stimulus;” “US” = “unconditioned stimulus”). ©2014 eLife Sciences Publications Ltd. Reproduced from [44] under Creative Commons attribution license.
And Now, the Cold Water
I. Toxicity?
As we have seen, evidence is emerging consistent with optimism that pharmacological modulation of ER stress and the ISR might be a valuable neuroprotective strategy, offering a potential bridge between the emergence of neuronal aggregate-clearing immunotherapies and the availability of effective cell therapy for neuronal replacement and reinforcement of circuitry. But having reviewed multiple reports of the benefits of inhibiting UPR/ISR signaling through eIF2α-P in animal models of neurodegeneration[31,34,37,39] and even normal cognitive and behavioral function,[44] it bears reminding ourselves that this pathway is ultimately an adaptive response that is activated by the cell in response to a range of stressors. During ER stress (via the UPR) or oxidative stress, hypoxia, viral replication, and nutrient insufficiency (via ISR)), eIF2α-P signaling acts to restore cellular homeostasis by simultaneously augmenting the cell’s protein quality-control machinery (thereby reducing the burden of unfolded and otherwise damaged proteins) and pausing the production of additional proteinaceous substrate (thereby preventing the further overloading of a taxed system) until the cell can once again manage the burden and normal protein translation and maturation resume. Alternatively, should these complementary responses be insufficient to allow the cell to resume normal functioning under the burden of unfolded or otherwise-damaged proteins, this same signaling pathway provides a means to dispose of the malfunctioning cell cleanly through apoptosis, abrogating the more deleterious necrotic destruction of the overwhelmed cell.[28,49]
One can anticipate, then, the potential maladaptive effects of pharmacological interference with UPR/ISR signaling, particularly if effective treatment requires chronic dosing. Sidrauski et al state that ISRIB “exhibits no overt toxicity in mice,”[44] but give no information on their basis for this summation: their published in vivo study involves experiments of less than one week’s duration, and gives no data on (for example) body weight, non-mnemonic functional tests, liver enzymes, serum creatine kinase, creatinine, or other standard means of monitoring even acute toxicity. And this study tested the effects on memory in young (in fact, weanling (6–7 wk old)), cognitively-intact animals without neurodegenerative disease, and therefore without the ER stress that such a drug might be used to help aging neurons bridge. In vitro work supporting the blockage of eIF2α signaling as the mechanism of action of ISRIB demonstrated that, by preventing the “escape valve” of ISR, this agent drives cells subjected to ER stress in culture to apoptose:
cells homozygous for non-phosphorylatable eIF2α, eIF2α(S51A), are unable to copewith ER stress properly, leading to reduced viability. This indicates that events downstream of eIF2α phosphorylation are required to resolve the stress. … ISRIB treatment of wild-type cells had similar consequences. Importantly, addition of ISRIB alone did not affect cell viability, as judged by the number of colonies that form after acute treatment. By contrast, ISRIB addition caused a strong synergistic effect on ER-stressed cells, reducing colony number and size significantly more than ER-stress alone [Figure 5 below]. This reduction in cell survival resulted from activation of apoptosis as the activity of the executioner caspases 3 and/or 7 was significantly induced under these conditions … The notion that ER stress remains unmitigated in ISRIB-treated cells is supported by sustained activation of all three UPR sensors [PERK, IRE1, and ATF6] … Importantly, in the absence of ER stress ISRIB treatment alone did not induce any of these sensors[44]
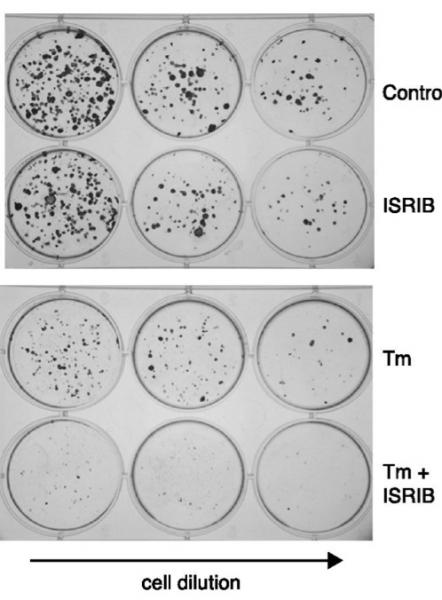
Figure 5. Pharmacological Suppression of UPR/ISR Enhances Apoptosis Under ER Stress. Human embryonic kidney cells with or without ISRIB under control (top) and ER-stressed (tunicamycin-treated) (bottom) conditions. ©2014 eLife Sciences Publications Ltd. Reproduced from [44] under Creative Commons attribution license.
Hints of the effects of ongoing UPR inhibition can be seen in the slightly more detailed information presented in Moreno et al‘s report on the effects of GSK2606414 on prion-infected mice.[34] Although little evidence on toxicology or animal health generally is presented, treated groups rapidly lost body weight, beginning almost immediately upon treatment initiation and continuing over the course of the brief 3-5 week period of testing. At the end of the study, prion-infected animals treated with vehicle had maintained their starting weights or possibly undergone a very slight weight loss, whereas animals treated with the drug had lost ≈20% of initial body weight, with no sign of a plateau having been reached (Figure S2). As a result, the investigators were mandated by British animal welfare regulations to euthanize their colony, leading to the perverse outcome that they were unable to perform survival analyses.[34] Aside from leaving a gap in our understanding of longer-term health and survival effects, the sheer pace of weight loss in treated groups suggests substantial toxicity to the drug in prion-infected animals.
Surprisingly, however, no such weight loss was observed in animals not infected with pathological prions. While no mechanistic explanation for the weight loss or its selectivity for infected animals was explored, the worrying implication is that the reason underlying the weight loss is not a generic toxicity specific to this agent, but is intimately involved with the drug’s mechanism of action in interfering with the UPR, which manifests precisely when the organism is subjected to elevated ER stress — i.e., in the target population for any future UPR-inhibiting neuroprotective drug.
There is greater biological insight on the hyperglycemia exhibited by their GSK2606414-treated mice, whose blood glucose levels climbed to 10-15 mM over the course of the experiment, vs. 8-10 mM in vehicle-treated controls.[34] While still below the range considered to indicate diabetes in mice (>22 mM), it is worrying to see so dramatic and treatment-specific a rise over the course of just 3-5 weeks, and other aspects of glucoregulation were not evaluated. In this case, there is a clear candidate explanation for this loss of glycemic control, and it is specific to the drug’s interference with the resolution of ER stress via the UPR. The pancreatic β-cell is subject to periods of intense secretory activity to meet the metabolic demands of meeting the acute postprandial rise in blood glucose with a commensurate burst of insulin secretion. The synthetic demand placed on the ER is very high, and the UPR is essential to maintaining β-cell homeostasis and viability in the face of the ensuing ER stress.[45-47] Moreover, the UPR and wider ISR are essential to meeting wider metabolic demands of fasting and feeding, including tissue insulin sensitivity, survival of glucagon-secreting pancreatic α-cells, and hepatic glucose output.[46,47]
The importance of the UPR/ISR in resolving this recurring cellular crisis can be seen in mice harboring mutations on the eIF2α phosphorylation site. Homozygous mice exhibit defective gluconeogenesis when fed normal chow, while heterozygous mutants fed a diabetogenic high-fat/high-sugar diet gain more weight, have a more adipose body composition, and suffer a more severe diabetic phenotype than wild-type mice on the same diet.[48] Conversely, liver-specific overexpression of the eIF2α-P phosphatase GADD34 was protective against diabetes and hepatosteatosis in animals fed a similar diet; when such animals are fed a normal diet, they remain lean, but suffer substantially reduced hepatic glycogen reserves and blunted gluconeogenesis, leading to susceptibility to hypoglycemia during fasting periods (reviewed in [46]). It is notable that lentiviral overexpression of this same phosphatase was found to be protective against neuropathology and cognitive and behavioral deficits in prion-infected mice[31]. Additionally, mice with elements of the UPR pathway knocked out exhibit a range of defects in skeletal, cartilaginous, and muscular development (reviewed in [49]).
Because our aim is to inhibit UPR/ISR signaling as an approach to the development of neuroprotective drugs, one potential approach to avoiding many of these toxic effects while still reaping neuroprotective benefits might be to design agents that selectively target the central nervous system (CNS) so as to avoid adverse effects on peripheral tissues. The favorable-looking effects of inhibiting UPR/ISR signaling on behavior and cognition in models of neurodegenerative disease[31,34,37,39] that have been the focus of this blog post, as well as prior work in disease models,[41-42,51] obviously bode well for such agents if peripheral toxicity is eliminated. Equally, the effects of ISRIB on cognitive function were reported to be entirely benign in short-term testing in normal juvenile control mice,[44] and GSK2606414 treatment had no deleterious effect on neuronal survival, hippocampal morphology, or burrowing behavior in uninfected control mice.[34]
However, other reports uncover the potential for a dark Janus face to even the prima facie beneficial effects of UPR inhibition on learning and memory, with learned behaviors becoming so readily and firmly ingrained as to impair cognitive/behavioral flexibility. Mice with forebrain-specific conditional knockout of PERK (PERK cKO mice) similar to the PERK-knockout control animals in the AD mouse study[37] exhibit several such defects.[40] When WT mice are exposed to an initial, preparatory weak stimulus, they are less prone to develop a conditioned startle response when a more abrupt stimulus is subsequently administered (so-called prepulse inhibition). This initial stimulus fails to protect PERK cKO mice from the startle response, implying impaired capacity for sensorimotor gating.[40] PERK cKO mice also exhibit exaggerated preference for a previously-familiarized object over a novel one in the novel object recognition task, behavioral perseveration when the hidden platform is relocated in the Morris water maze and Y-water maze, and impaired extinction of fear-based learning (Figure 6).[40] These effects were reversed by treatment with the glycine transporter inhibitor SSR504734, which also restored eIF2α-P and expression of ATF4 in PERK cKO mice.[40]

Figure 6. PERK knockout impairs the ability of mice to adapt to new conditions by “unlearning” previously-learned cues. Reproduced from [40]. © 2012 the Authors.
And while the focus of this blog post is on the cognitive effects of inhibiting UPR/ISR, there is evidence that doing so may dysregulate brain functions other than learning and memory. For example, ER stress is involved in central responses to insulin and leptin signaling and inflammation,[46,59,60,61] raising the possibility that chronic pharmacological modulation of UPR/ISR signaling may lead to dysregulated energy balance.
II. Effects on the Wider ER Stress response
The drugs and mutations discussed to date have targeted signaling via phosphorylation of eIF2α by PERK and other means, and have largely neglected the other pathways through which the cell responds to ER stress. It is common practice to treat these various arms of the ER stress response as largely independent, parallel pathways that proceed independently once activated by the upstream ER stress sensors (Figure 7). However, several recent studies have found evidence of substantial crosstalk amongst the various ER stress pathways, which as we shall see has implications for the therapeutic use of agents that might otherwise be throught to be specific inhibitors of UPR/ISR signaling via eIF2α-P.
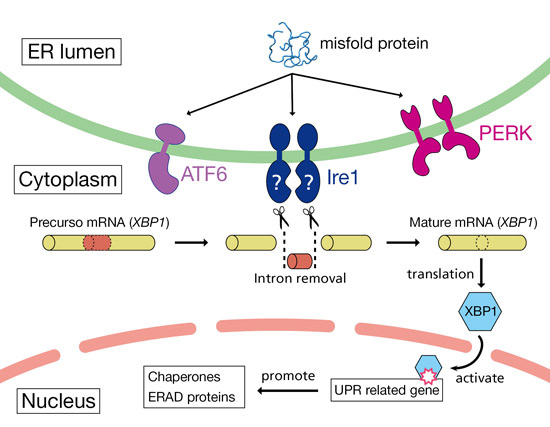
Figure 7. Canonical parallelism of aspects of ER stress response. In response to ER stress, IRE1 exonuclease removes the intron from XBP1 pre-mRNA, leading to splicing and translation of mature XBP1. XBP1 protein then upregulates the expression of the chaperones and proteins involved in ERAD. Reproduced from Protein Data Bank Japan (PDBj) under public domain license.
In 2011, for instance, Teske et al found that genetic abrogation of signaling through eIF2α-P also inhibits the ATF6 and IRE1/XBP1 pathways. Under conditions of ER stress, removal of components of the ISR either immediately upstream (PERK) or downstream (ATF4) of eIF2α-P reduces translation and induction of ATF6, and also reduces expression of XBP1, despite an apparent lack of effect on IRE1 cleavage of XBP1 precursor mRNA (Figure 7).[64] “As a consequence, liver-specific depletion of PERK significantly reduces both the translational and transcriptional phases of the UPR, leading to reduced protein chaperone expression, disruptions of lipid metabolism, and enhanced apoptosis.”[64] Later studies revealed even more linkages within what are generally regarded as separate pathways within the UPR, as the phosphorylated (active) form of ATF6 is produced in cells undergoing ER stress prior to the phosphorylated (active) form of XBP1, and the two form a heterodimer that transactivates and transcriptionally induces several ERAD chaperones.[71]
This has troubling implications for pharmacological inhibition of UPR/ISR as a neuroprotective strategy, as overexpression of XBP1 is neuroprotective in vivo in Drosophila retinal neurons overproducing Aβ1-42, whereas partial knockdown of the gene enhances neurotoxicity; these effects can be recapitulated in PC12 cells treated with Aβ oligomers.[65 (and see (62)] More recently, several polymorphisms of XBP1 have been found to be associated with higher risk of AD and with faster cognitive decline in established AD in a Chinese Han population.[63] Thus, agents inhibiting ISR/UPR signaling through eIF2α might indirectly enhance Aβ toxicity by interfering with other aspects of the ER stress response.
Translatability to the Aging ± Rejuvenating Population
I. Contrast with Extreme Phenotype in Neurodegenerative Models
The animal studies reviewed heretofore have been conducted in animals at two extremes of the neurodegenerative continuum: on the one hand, mice and flies with severe, rapid-onset protein misfolding diseases[31,34,37,39] and on the other hand, juvenile WT mice presumptively free of neuropathology or chronic ER stress.[44] But the target population for the neuroprotective agents posited for this blog post can be projected to be situated somewhere between these two extremes, with variations in ER stress varying from one individual to the next, and from one brain region to the next, and at different phases in their brain rejuvenation cycles. There is reason to believe that these differences between the model and the modeled may in several ways give a misleading impression of the neuroprotective potential of UPR-inhibiting agents.
Rate of Protein Synthesis
The transgenic mouse models of neurodegenerative aging processes used in the studies reviewed here utilize overexpression of transgenic human wild-type or, more often, strongly disease-associated mutated versions of the proteinaceous substrates of intraneuronal aggregates whose accumulation drives particular brain aging phenotypes. In “normal” human aging, of course, the vast majority of persons’ neurons are accumulating proteinaceous aggregates as a result of wild-type proteins being synthesized at normal rates dictated by normal physiological regulatory systems. And while future aggregate-clearing rejuvenation biotechnologies will remove existing misfolded proteins from the neuron, it is a core feature of the “damage-repair” heuristic that these therapies will not interfere with the metabolic determinants of their formation, such as rate of synthesis of the native protein or the various factors that impact its subsequent aggregation. Thus, while the level of proteinaceous aggregates present in the neurons of the first recipients of such therapies will be reduced relative to similarly-aged persons receiving no such therapies, the rate of new precursor synthesis, passage through the ER, and aggregation will be unaffected (except inasmuch as aggregate clearance and other structural restoration will abrogate vicious metabolic cycles (of oxidative and inflammatory stress, e.g.) introduced by advanced neurodegenerative processes).
The reason this contrast may be important in projecting the potential translatability of the effects of UPR-inhibiting drugs observed in models of neurodegenerative aging process is that the core function of rough ER is to ensure the native folding and transport of newly-synthesized proteins. Conditions of high protein synthetic demand are thus of themselves a source of ER stress. This is the reason why (for example) pancreatic β-cells are subject to acute ER stress as insulin secretory demand ramps up to meet the postprandial rise in blood glucose, making the UPR critical to maintaining β-cell homeostasis and viability.[45-47] Thus, the observed benefits of UPR-inhibiting drugs in these models may be in part an artifactual “correction” of the ER stress generated by the forced overexpresssion utilized in creating these animal models that is not present in the neurons of normal aging humans or mice.
Locus and Mechanism of Aggregation
Moreover, while excessive production of proteins leads to their misfolding in the ER through the exceeding ER protein-folding capacity, the main determinants of formation of the aggregates driving neurodegenerative aging process occur in several other cellular compartments (including the cytosol, the endosome, and the lysosome), as well as “infectious” from cell-to-cell by templating and transsynaptic spread[72]). They are also driven by multiple enzymatic and nonenzymatic factors unrelated to the sheer quantity of protein being synthesized at a given time or present at steady state. It is reasonable to ask whether aggregation that occurs outside of the ER and that either forms stable cytosolic inclusions or is targeted by the autophagic machinery would be as prone to induce ER stress as aggregates arising in large part within the ER itself. If not, then the transgenic mouse models of neurodegenerative disease would again inflate the contribution of ER stress to “normal” age-related neurodegeneration, and proportionately inflate the potential benefit of pharmacologically suppressing the UPR that it elicits.
Involvement of Multiple Aggregates
While transgenic animal models of neurodegenerative aging processes only introduce one or sometimes two ER-inducing aggregates into the neuron, the aging brain concomitantly accumulates numerous intracellular aggregates. Under the baseline scenario of “aging as usual,” each such aggregate accumulates at varying rates from individual to individual at a given age based on a range of genetic, environmental, and lifestyle factors, and each of these aggregates accumulates differentially in different anatomical regions of the brain. The level of UPR inhibition that might best protect against synaptic toxicity and neuronal dysfunction driven by Aβ in a given individual will not necessarily be optimal for protecting against tau neurotoxicity or α-synuclein aggregates in the same individual, within the same neuron, or across different brain regions — and all of these pharmacodynamic relationships will further vary from one individual to the next. Thus, the a dose that is highly protective in one area of the brain for one individual may be alternatively ineffective or neurotoxic in another.
We will here highlight relevant research as regards the two most important such aggregates: pathological tau species (involved in AD and in frontotemporal dementia and other tauopathies) and aggregated α-synuclein (involved in PD, particularly in the nonmotor symptoms of the disease).
a. Tau Pathology
Unlike in human AD, Aβ overproduction in the transgenic mice used in Ma et al‘s transgenic AD mouse study[37] does not result in downstream tau pathology. In addition to being uninformative about this critical aspect of the aetiopathogenesis of age-related cognitive decline and clinical AD, there is some reason for concern that inhibition of the UPR/ISR might lead to unopposed neurotoxicity from pathogenic tau. As noted previously, induction of the UPR is an early event in both AD[22-24] and genetic- and age-related tauopathies.[24,27,66] (see [28] for a recent review). Notably, in one study including both tauopathy and AD subjects,[66] activation of the UPR was highly linked to tau neuropathology: tauopathy subjects exhibited activation of PERK and PKR “in all analyzed brain areas that contained abnormal tau protein. Unlike AD, immunoreactivity for both enzymes was, like that for phospho-tau, present not only in neurons, but also in astrocytes and oligodendrocytes … [with] high-grade colocalization of phospho-PERK and PKR with phospho-tau”.
Evidence also exists to suggest that this tau-associated induction of UPR is neuroprotective against tau pathology. In Drosophila, transgenes expresssing human wild-type tau, FTDP-17-linked tauR406, and tau with disease-associated mutations at multiple phosphorylation sites are progressively more neurotoxic, and neurons exhibit parallel progressive increases in XBP1 protein levels.[65] That XBP1 serves a neuroprotective function is suggested by the observation that either heterozygous XBP1 loss-of-function mutations or anti-XBP1 silencing RNA roughly doubled the rates of neuronal apoptosis in flies with human tau transgenes over and above the levels in the same animals with intact XBP1, whereas neither silencing nor mutation of XBP1 was significantly harmful to otherwise wild-type Drosophila neurons.[65] Consistent with this, genetic variants in EIF2AK3 (the gene encoding PERK) have been associated with elevated risk of progressive supranuclear palsy (PSP), a rare tauopathy.[67] Thus, the report of beneficial effects of pharmacological inhibition of PERK activity in animal models of AD with no involvement of tau[37] or that otherwise lack tau pathology[31,37,39,44] might be misleading in terms of overall effects on cognitive function or disease progression, due to a failure to capture the full scope of effects on Aβ vs. tau pathology vs. acute memory formation vs. neuronal survival.
b. α-Synuclein
AS aggregates are the third major class of aggregate commonly found concomitantly present with Aβ and pathological tau species in the aging human brain. As previously noted, signs of UPR activation and ER stress are increased early in PD,[23,25,26] with phosphorylated PERK colocalizing with increased AS in substantia nigra (SN) dopaminergic neurons.[25] But even considering AS aggregates in isolation, predicting the conditions under which the induction or suppression of the UPR will exhibit neuroprotective vs. neurotoxic effects seems especially difficult. In one standard animal model of PD, administration of the dopaminergic (DA) neuron-selective mitochondrial toxin 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) leads to the production of ubiquitinated inclusion bodies in DA neurons of young adult mice, accompanied by neuronal loss. The surviving neurons exhibited activated ATF6 and upregulation of ER chaperones, while conversely ATF6 knockout mice exhibited enhanced MPTP neurotoxicity, demonstrating the importance of UPR induction in neuronal survival in this model.[68]
Yet surprisingly, when researchers at the Biomedical Neuroscience Institute at the University of Chile administered the selective DA neurotoxin 6-hydroxydopamine (6-OHDA) to mice, the ensuing loss of DA neurons in the SN is substantially reduced in specific SN subregions in animals with conditional, neuron-specific knockout of XBP1.[69] Probing this result further, the investigators discovered that under basal conditions, these animals’ DA neurons exhibit an increased level of aggregated AS, and large intraneuronal inclusions with an electrophoretic pattern similar to that observed in model mice harboring the PD-associated A53T α-synuclein mutation. Alongside these inclusions, unchallenged DA neurons exhibit a modest increment in basal ER stress and evidence of induction of autophagy, but no increase in CHOP expression or obvious loss of neurons.[69]
The Chilean investigators hypothesized that in these animals, lifelong lack of XBP1 leads to chronic low-level, unresolved ER stress, caused and/or resulting in the accumulation of low (rather than nonexistant) levels of AS inclusions. The ensuing chronic compensatory induction of other constituents of the UPR then constitute a form of ER “preconditioning” that increases the ability of DA neurons to survive under the more severe insult of 6-OHDA.[69] In support of this hypothesis, a previous study headed by Bertrand Mollereau of the Laboratoire de Biologie Moléculaire de la Cellule in Lyon, France demonstrated that pharmacological “preconditioning” of 6-OHDA-treated mice with low doses of the UPR-inducing drug tunicamycin afforded substantial neuroprotection to DA neurons (Figure 8A), accompanied by reduced frequency of rotational behaviors under dopaminergic agonism following unihemispheric DA neuronal loss (Figure 8B).[70] The same French study found similar results in transgenic Drosophila overexpressing the human AS protein in DA neurons, and in human SH-SY5Y neuroblastoma cells exposed to 6-OHDA.[70]
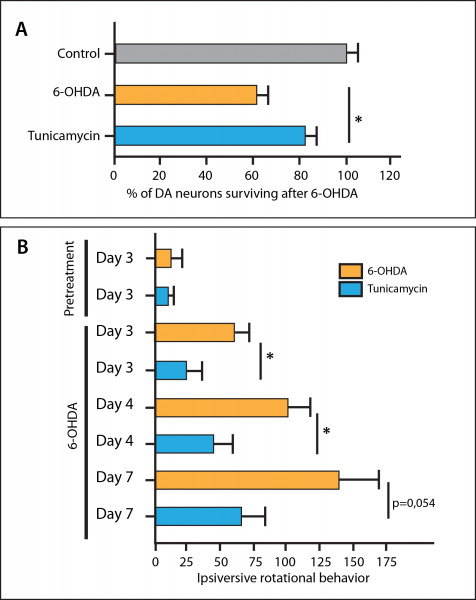
Figure 8. Preconditioning ER stress reduces substantia nigra dopaminergic neuron loss (A) and ameliorates functional deficits (B) in mice treated with from dopaminergic neurotoxin. *p ≤ 0.05. Redrawn by Anne Corwin from [70].
Consistent with the neuroprotective effects of the UPR against AS in general and the “preconditioning” model in particular, the Chilean group found that knockdown of XPB1 in the SN of wild-type adult animals led to an increase in markers of ER stress, increased CHOP mRNA, and loss of dopaminergic neurons, with surviving cells exhibiting abnormal morphology and atrophic dendrites and axons, and a loss of innervation of the striatum by DA neurons. Conversely, stereotactic delivery of XBP1 transgenes to mouse SN induced the expression of several UPR target genes, following 6-OHDA insult the treated hemispheres exhibited a ≈42% reduction in striatal DA neuronal loss and reduced loss of striatal innervation.[69]
Beyond the specific results for AS neuropathology, or the complicating factor of the coexistence and regional variation of multiple additional aggregates in neurons located in different regions of the brain, the results from the Chilean[69] and French[70] ER stress preconditioning studies clearly sound a note of caution on UPR inhibition per se as a neuroprotective strategy.
II. Suggestion of Deleterious Effects in "Normal" Brain Aging
To repeat: the target population for any potential UPR-inhibiting neuroprotective agents will be persons whose aging neurons are subject to a substantially higher level of ER than are those of the young wild-type control animals used in these studies,[34,44] and yet substantially lower level than in animals suffering the aggressive amyloidosis of RML prion disease[31,34] or severe overproduction of Aβ[37] or TDP-43.[39] In addition to the differing level of ER stress, the mechanisms and locus of protein aggregation, as well as the number and regional variation in types of aggregate, present in these models may vary substantially from those in aging and partially-rejuvenated brains. An agent intended to serve as a neuroprotective bridge toward robust cell replacement therapy will need to cantilever such persons out into an ambiguous space in a period during which metabolism continues to produce the various proteinaceous aggregates involved in the neurodegenerative diseases of aging, but their abundance is kept to a range below the “threshold of pathology” through periodic therapeutic clearance.
No direct and intentional attempt to model the effects of UPR inhibition in the CNS of such persons have been performed. However, there are suggestions of a deleterious effect of UPR inhibition in some aspects of cognitive function in one of the control groups in Ma et al‘s model AD study[37]. To test the effects of late-developmental hippocampus-selective PERK knockout on aging AD-transgenic mice, these investigators rigorously compared such animals to three control groups of age-matched animals: purely WT mice; AD-transgenic mice receiving no treatment; and otherwise-WT aging animals with the same PERK knockout as the “treated” AD mice.
It is this last group that is most analogous to aging humans subjected to a strong UPR-inhibiting drug. These animals exhibited better performance on some cognitive tests than did untreated AD mice. But notably, perhaps, their performance was nonsignificantly poorer on some tests than either AD mice “treated” with the same knockout of PERK, or purely WT aging mice (see Figure 2(c,e) and just possibly (b)).[37] While one must be cautious in taking at face value raw numerical differences that did not reach statistical significance, the apparent presence of cognitive deficits in aging PERK knockout mice is consistent with a model in which inhibition of PERK is beneficial in the brains of animals (or persons) subjected to very severe ER stress, but somewhat harmful in animals subjected to a more moderate level of ER stress typical of “normal” aging. Such a biphasic effect is consistent with the very biological function of the UPR, as a pathway whose primary purpose is to restore homeostasis in neurons subjected to ER stress.[28,49] A similar situation might prevail in recipients of aggregate-clearing rejuvenation biotechnologies, in whom levels of aggregates are held consistently below the threshold of pathology but none the less vary between very low and moderately high levels depending on the periodicity of rounds of treatment.
The positive effects of PERK knockout on cognitive function in Ma et al‘s transgenic AD mouse study[37] are much less informative than they might seem, even under the more extreme ER stress of frank diagnosable AD or its late preclinical stages. The APPswe/PS1ΔE9 mutations used to induce AD-like neuropathology and phenotypes in these mice lead to massive overproduction of Aβ, but do not result in substantial neuronal loss. Thus, the ability (or lack of it) of PERK inhibition to actually enhance neuronal survival downstream of Aβ accumulation remains untested. If anything, there was some suggestion in this study that PERK inhibition might not be sufficient to prevent UPR-driven apoptosis in the AD brain: in these AD model mice, unlike in the prion-infected mice used in Moreno et al‘s studies,[31,34] blocking the PERK/eIF2α arm of the UPR did not normalize the elevated levels of CHOP in the hippocampus.[37]
At the risk of stating the obvious, it is imperative to perform adequately-powered studies extensively testing the cognitive and other effects of ISRIB, GSK2606414, and other UPR/ISR inhibiting agents in normally-aging mice before they are advanced into human testing as neuroprotectants, cancer therapies, or for any other indication.
The Interface with Aggregate-Clearing Rejuvenation Biotechnology
As we have reviewed, many of the differences between the actual process of human neurodegenerative aging and the animal models of particular neurodegenerative disease in which UPR inhibitors have been tested as neuroprotectants suggest some downward revaluation of the potential clinical benefit of this approach. Most or all of these reasons for caution appear likely to apply equally to people undergoing “aging as usual” as to future beneficiaries of periodic aggregate-clearing rejuvenation biotechnologies. But therapeutic removal of accumulated aggregates from aging neurons may also change the therapeutic context in ways that make effective use of these drugs as neuroprotectants even more difficult and limited than in “normal” aging.
The neurons of both transgenic animal models of age-related neurodegeneration and of persons undergoing actual and unmitigated neurodegenerative aging processes suffer from either a continuous, high burden of intra- and extraneuronal aggregates, or from a more or less rapidly increasing such burden. Experimental studies suggest that UPR-inhibiting drugs might preserve the function of neurons that would otherwise be disabled by the UPR’s initiation of general translational arrest, or pushed into apoptosis. However, there must clearly exist some threshold of pathology beyond which the burden of such damage will exceed the ability of UPR inhibition to rescue neurons.
Recipients of future rejuvenation biotechnologies would presumably enjoy a substantial expansion of the percentage of potentially salvageable neurons in their brains, thanks to the periodic removal of such aggregates. But unlike the animal models or “normal” brain aging (manifest in specific, diagnosed neurodegenerative disease or not), the neurons of the first recipients of these therapies would be periodically cleared of aggregates by immunotherapy, leading to oscillations between lower and higher burdens of each aggregate and of their sum, both within individual neurons and amongst different neuronal populations.
The range between a neuron resident in the brain of a recent recipient of aggressive application of a given aggregate-clearing therapy and one who is at the end of such a cycle and due for another could be minimal or quite substantial, depending on risk-to-benefit profile and whether an active vaccine (long-term, gradually waning clearance) or passive immunotherapy (short-term, reversible, more frequently administered clearance) is used. And especially in the case of the early iterations of such therapies, the degree of thoroughness with which a given clearance therapy can safely clear its target from the brain as a whole or in specific vulnerable neuronal populations will certainly be suboptimal for each, and vary from one aggregate to another. Moreover, the rate at which such aggregates will resume accumulation after a given round of therapeutic “reset” will continue to vary based on the same range of genetic, lifestyle, and environmental factors that cause variations in accumulation rates in “normal” aging.
Each of thes considerations will be expected to introduce further divergence in the intra-individual optima for protecting different neuronal populations in the face of multiple ER stressors. In principle, the level of drug necessary to triage the survival of the maximum number of neurons could be titrated to individuals based on biomarkers, imaging, or functional tests to reveal response to therapy, within the tolerable limits of side-effects. But in the early years of use of aggregate-clearing rejuvenation biotechnologies, clinical experience with each of the agents involved — the individual therapeutics used to clear each of the several target aggregates, as well as the UPR-inhibiting neuroprotectant — will be minimal. Because all age-related aggregate accretions are ultimately deleterious, the optimum level of each is presumptively zero, and the zealousness with which aggregate-clearing therapies should be applied is therefore limited only by considerations of safety and therapeutic diminishing returns. Therefore, adjusting the individual patient’s dose and timing of individual aggregate-clearing in isolation may not be difficult, and using them in combination only modestly more so.
But to perform the combinatorial optimization required to tune the level of UPR activity to ER stress “tone” in the presence of several different aggregates, being produced at different rates and then cleared to varying degrees by future rejuvenation biotechnologies on different schedules, is evidently a very daunting prospect, making the likely risk-to-benefit ratio of UPR inhibition agents as bridging neuroprotectors even murkier.
A Promising Candidate — And the Need for a Wider Field
From what we have reviewed, it is far from clear whether the therapeutic dilemmas involved in adjusting UPR activity to avert neurotoxicity in individual patients, suffering with varying burdens and rates of accumulation of a range of aggregates differentially affecting different regions of the brain, can be overcome to realize the neuroprotective potential of this approach demonstrated in animal models of neurodegenerative disease.[31,37,39,44] Were a clinically effective neuroprotective agent to emerge in the next 5 years, its efficacy at preserving remaining neurons at any given time would necessarily diminish over time, and likely at an accelerating rate, due to the progressive and synergistic nature of the processes underlying degenerative aging in the brain and elsewhere across the body. In the face of ongoing UPR-inhibiting neuroprotective treatment, the contributors to ER stress would continue to become more severe, as aging neurons continue to suffer with progressively greater accumulations of the hallmark neuropathologies of AD and PD. Additionally, both they and the cells and stroma with which they interact would also be increasingly burdened with other forms of age-related cellular and molecular damage. Their capacity to maintain function and viability would be further constrained by a systemic milieu that is increasingly deranged by the downstream metabolic consequences of degenerative processes in the local environment and at remote locations across the body.[73-77] As with much of geriatric medicine today, clinical futility would rapidly be reached, as would in many cases a point beyond which the side-effects of the drug clearly outweigh any benefits.
By contrast, neuroprotective therapies might prove extremely valuable in the scenario envisioned in this blog post, in which a partial panel of rejuvenation biotechnologies are available to periodically remove, repair, replace, or render harmless some significant number of such lesions, but cell therapy for the brain has not yet matured. At any given calendar age, the brain of the recipient of such therapies can be expected to contain a larger number of neurons with sufficient remaining viability as to be salvageable by neuroprotective therapies, and periodic rounds of rejuvenation would extend and periodically resuscitate the neuroprotectant’s effectiveness by interrupting or reversing the downward pull of degenerative aging processes. But as we have seen, there may be additional sources of potential therapeutic dilemma that hinder or even preclude the effective clinical use of UPR-inhibiting drugs as a neuroprotective strategy in this context.
To date, despite extensive efforts to repurpose existing drugs to protect aging neurons from neurodegenerative processes[13-15,30] and to develop novel rationally-designed neuroprotectants[16-21,50] no neuroprotective therapies have emerged. Efforts to identify such should certainly continue, both for the hopes patients and their loved ones who suffer today or are at near-term risk of clinical neurodegenerative disease, and to serve the bridging function for the first recipients of rejuvenation biotechnologies for the brain that this blog post envisions. But in doing so, it must be understood that even if initially successful, the potential benefit of any neuroprotective agent based on the modulation of metabolic determinants of the cellular and molecular damage of aging in the brain, or of the downstream metabolic consequences of such damage will necessarily be limited in scope and in time by ongoing degenerative aging processes.
Durable intervention in aging humans must target the underlying structural damage of aging tissues directly — in this case, through the development of mature neuronal replacement therapy to reinforce and rebuild damaged neural circuitry, to complement the more imminent arrival of aggregate-clearing rejuvenation biotechnologies. Happily, in the time between the reports that drove the recent upsurge in interest in UPR inhibition as a neuroprotective strategy [34,37] and the publication of this blog post, there have been several very promising reports of progress toward this goal. Neural stem cells derived from a stable human fetal cell line have been transplanted into the brains of nonhuman primates and survived for two years, without the use of immunosuppressants and without any tumor-risk-associated posttransplant replication.[78] Another group directly converted mouse fibroblasts into induced neural stem cells (iNSC) using a 5-factor retroviral cocktail of selected Yamanaka stem cell and neural-specific transcription factors, and then injected into the dentate gyrus of immunodeficient mice.[79] These cells survived at high levels for 6 months without any evidence of tumorigenic overgrowth of the graft site, and while a majority of them formed glial cells, substantial numbers of those located at the edges of the graft or that migrated outside of the graft zons underwent a migratory neuronal differentiation process similar to endogenous neural stem cells when migrating into the subgranular zone.[79] Critically, these iNSC-derived neurons integrated into the local neuronal networks, exhibiting similar morphology and orientation to the local endogenous neurons in the circuit, forming direct synaptophysin-positive synaptic connections with surrounding endogenous neurons, and exhibiting neuron-like electrophysiological properties.[79]
In the battle between medicine and disease, we should ever continue to embrace the best tools available at any given time, imperfect though they may be. There is certainly great potential for synergy between agents that modulate metabolic pathways to reduce the formation of aging damage and agents that remove it, especially relatively early on in the process of degenerative aging, and especially in the early days of rejuvenation biotechnology, when the scope and scale of damage for which regenerative therapies have been developed is limited. But our work will be incomplete, and we can never rest, until it is within our power to restore tissues to youthful health and function by direct removal, replacement, and reinforcement of cells and biomolecules worn by time. Only then may all of us reap and widely sow the fruits of humanity’s long scientific quest for long lives and robust health for all.
Notes
* However, evidence has recently emerged that ER stress leads to the induction of autophagy but not the proteasome in neurons, suggesting the lysosome as the preferred and perhaps the exclusive site of degradation of ER stressor proteins in age-related neurodegeneration.[29]
†The elevation of PSD-95 in transgenic AD mice seen in reference 37 and elsewhere is actually opposite in direction to the suppression of this protein in prion-infected mice;[34] however, the re-establishment of levels similar to control animals by inhibition of eIF2α phosphorylation is a commonality of the two experiments.
References
1: Doré V, Villemagne VL, Bourgeat P, Fripp J, Acosta O, Chetélat G, Zhou L, Martins R, Ellis KA, Masters CL, Ames D, Salvado O, Rowe CC. Cross-sectional and Longitudinal Analysis of the Relationship Between Aβ Deposition, Cortical Thickness, and Memory in Cognitively Unimpaired Individuals and in Alzheimer Disease. JAMA Neurol. 2013 Jul 1;70(7):903-11. doi: 10.1001/jamaneurol.2013.1062. PubMed PMID: 23712469.
2: Doi D, Morizane A, Kikuchi T, Onoe H, Hayashi T, Kawasaki T, Motono M, Sasai Y, Saiki H, Gomi M, Yoshikawa T, Hayashi H, Shinoyama M, Refaat MM, Suemori H, Miyamoto S, Takahashi J. Prolonged maturation culture favors a reduction in the tumorigenicity and the dopaminergic function of human ESC-derived neural cells in a primate model of Parkinson’s disease. Stem Cells. 2012 May;30(5):935-45. doi: 10.1002/stem.1060. PubMed PMID: 22328536.
3: Barker RA, Barrett J, Mason SL, Björklund A. Fetal dopaminergic transplantation trials and the future of neural grafting in Parkinson’s disease. Lancet Neurol. 2013 Jan;12(1):84-91. doi: 10.1016/S1474-4422(12)70295-8. Review. PubMed PMID: 23237903.
4: Redmond DE Jr, Weiss S, Elsworth JD, Roth RH, Wakeman DR, Bjugstad KB, Collier TJ, Blanchard BC, Teng YD, Synder EY, Sladek JR Jr. Cellular repair in the parkinsonian nonhuman primate brain. Rejuvenation Res. 2010 Apr-Jun;13(2-3):188-94. Review. PubMed PMID: 20370501; PubMed Central PMCID: PMC2946058.
5: Diaz F, McKeehan N, Kang W, Hébert JM. Apoptosis of glutamatergic neurons fails to trigger a neurogenic response in the adult neocortex. J Neurosci. 2013 Apr 10;33(15):6278-84. doi: 10.1523/JNEUROSCI.5885-12.2013. PubMed PMID: 23575827; PubMed Central PMCID: PMC3644398.
6: Lindvall O, Kokaia Z. Stem cells in human neurodegenerative disorders–time for clinical translation? J Clin Invest. 2010 Jan;120(1):29-40. doi: 10.1172/JCI40543. Review. PubMed PMID: 20051634; PubMed Central PMCID: PMC2798697.
7: Gopurappilly R, Pal R, Mamidi MK, Dey S, Bhonde R, Das AK. Stem cells in stroke repair: current success and future prospects. CNS Neurol Disord Drug Targets. 2011 Sep 1;10(6):741-56. Review. PubMed PMID: 21838668.
8: Xu X, Warrington AE, Bieber AJ, Rodriguez M. Enhancing CNS repair in neurological disease: challenges arising from neurodegeneration and rewiring of the network. CNS Drugs. 2011 Jul;25(7):555-73. doi: 10.2165/11587830-000000000-00000. Review. PubMed PMID: 21699269; PubMed Central PMCID: PMC3140701.
9: McKeehan N, Diaz F, Kang W, Hébert JM. What good is an old brain in a young body – a strategy for regenerating the neocortex. Abstracts of Strategies for Engineered Negligible Senescence (SENS), Sixth Conference. Cambridge, United Kingdom. September 3–7, 2013. Rejuvenation Res. 2013 Sep;16 Suppl 1:S24 (Abs 40). doi: 10.1089/rej.2013.16.sens6abs. PubMed PMID: 23944655.
10: Kernie SG, Parent JM. Forebrain neurogenesis after focal Ischemic and traumatic brain injury. Neurobiol Dis. 2010 Feb;37(2):267-74. doi: 10.1016/j.nbd.2009.11.002. Epub 2009 Nov 10. Review. PubMed PMID: 19909815; PubMed Central PMCID: PMC2864918.
11: van Strien ME, van den Berge SA, Hol EM. Migrating neuroblasts in the adult human brain: a stream reduced to a trickle. Cell Res. 2011 Nov;21(11):1523-5. doi: 10.1038/cr.2011.101. Epub 2011 Jun 21. PubMed PMID: 21691300; PubMed Central PMCID: PMC3364722.
12: Bergmann O, Liebl J, Bernard S, Alkass K, Yeung MS, Steier P, Kutschera W, Johnson L, Landén M, Druid H, Spalding KL, Frisén J. The age of olfactory bulb neurons in humans. Neuron. 2012 May 24;74(4):634-9. doi: 10.1016/j.neuron.2012.03.030. PubMed PMID: 22632721.
13: Palmer AM. Neuroprotective therapeutics for Alzheimer’s disease: progress and prospects. Trends Pharmacol Sci. 2011 Mar;32(3):141-7. doi: 10.1016/j.tips.2010.12.007. Epub 2011 Jan 20. Review. PubMed PMID: 21256602.
14: Lipton SA. Paradigm shift in neuroprotection by NMDA receptor blockade: memantine and beyond. Nat Rev Drug Discov. 2006 Feb;5(2):160-70. Review. PubMed PMID: 16424917.
15: Parsons CG, Stöffler A, Danysz W. Memantine: a NMDA receptor antagonist that improves memory by restoration of homeostasis in the glutamatergic system–too little activation is bad, too much is even worse. Neuropharmacology. 2007 Nov;53(6):699-723. Epub 2007 Aug 10. Review. PubMed PMID: 17904591.
16: Nagahara AH, Merrill DA, Coppola G, Tsukada S, Schroeder BE, Shaked GM, Wang L, Blesch A, Kim A, Conner JM, Rockenstein E, Chao MV, Koo EH, Geschwind D, Masliah E, Chiba AA, Tuszynski MH. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat Med. 2009 Mar;15(3):331-7. doi: 10.1038/nm.1912. Epub 2009 Feb 8. PubMed PMID: 19198615; PubMed Central PMCID: PMC2838375.
17: Caraci F, Spampinato S, Sortino MA, Bosco P, Battaglia G, Bruno V, Drago F, Nicoletti F, Copani A. Dysfunction of TGF-β1 signaling in Alzheimer’s disease: perspectives for neuroprotection. Cell Tissue Res. 2012 Jan;347(1):291-301. doi: 10.1007/s00441-011-1230-6. Epub 2011 Aug 31. Review. PubMed PMID: 21879289.
18: Tripathy D, Thirumangalakudi L, Grammas P. RANTES upregulation in the Alzheimer’s disease brain: a possible neuroprotective role. Neurobiol Aging. 2010 Jan;31(1):8-16. doi: 10.1016/j.neurobiolaging.2008.03.009. Epub 2008 Apr 28. PubMed PMID: 18440671; PubMed Central PMCID: PMC2803489.
19: Counts SE, Perez SE, Ginsberg SD, Mufson EJ. Neuroprotective role for galanin in Alzheimer’s disease. EXS. 2010;102:143-62. Review. PubMed PMID: 21299067; PubMed Central PMCID: PMC3117305.
20: Pacher P, Szabo C. Role of the peroxynitrite-poly(ADP-ribose) polymerase pathway in human disease. Am J Pathol. 2008 Jul;173(1):2-13. doi: 10.2353/ajpath.2008.080019. Epub 2008 Jun 5. Review. PubMed PMID: 18535182; PubMed Central PMCID: PMC2438280.
21: Schapira AH, McDermott MP, Barone P, Comella CL, Albrecht S, Hsu HH, Massey DH, Mizuno Y, Poewe W, Rascol O, Marek K. Pramipexole in patients with early Parkinson’s disease (PROUD): a randomised delayed-start trial. Lancet Neurol. 2013 Aug;12(8):747-55. doi: 10.1016/S1474-4422(13)70117-0. Epub 2013 May 31. PubMed PMID: 23726851; PubMed Central PMCID: PMC3714436.
22: Hoozemans JJ, van Haastert ES, Nijholt DA, Rozemuller AJ, Eikelenboom P, Scheper W. The unfolded protein response is activated in pretangle neurons in Alzheimer’s disease hippocampus. Am J Pathol. 2009 Apr;174(4):1241-51. doi: 10.2353/ajpath.2009.080814. Epub 2009 Mar 5. PubMed PMID: 19264902; PubMed Central PMCID: PMC2671357.
23: Hoozemans JJ, van Haastert ES, Nijholt DA, Rozemuller AJ, Scheper W. Activation of the unfolded protein response is an early event in Alzheimer’s and Parkinson’s disease. Neurodegener Dis. 2012;10(1-4):212-5. doi: 10.1159/000334536. Epub 2012 Feb 1. Review. PubMed PMID: 22302012.
24: Stutzbach LD, Xie SX, Naj AC, Albin R, Gilman S; PSP Genetics Study Group, Lee VM, Trojanowski JQ, Devlin B, Schellenberg GD. The unfolded protein response is activated in disease-affected brain regions in progressive supranuclear palsy and Alzheimer’s disease. Acta Neuropathol Commun. 2013 Jul 6;1(1):31. doi: 10.1186/2051-5960-1-31. PubMed PMID: 24252572.
25: Hoozemans JJ, van Haastert ES, Eikelenboom P, de Vos RA, Rozemuller JM, Scheper W. Activation of the unfolded protein response in Parkinson’s disease. Biochem Biophys Res Commun. 2007 Mar 16;354(3):707-11. Epub 2007 Jan 17. PubMed PMID: 17254549.
26: Ferrer I, Martinez A, Blanco R, Dalfó E, Carmona M. Neuropathology of sporadic Parkinson disease before the appearance of parkinsonism: preclinical Parkinson disease. J Neural Transm. 2011 May;118(5):821-39. doi: 10.1007/s00702-010-0482-8. Epub 2010 Sep 23. Review. PubMed PMID: 20862500.
27: Nijholt DA, van Haastert ES, Rozemuller AJ, Scheper W, Hoozemans JJ. The unfolded protein response is associated with early tau pathology in the hippocampus of tauopathies. J Pathol. 2012 Apr;226(5):693-702. doi: 10.1002/path.3969. Epub 2012 Feb 17. PubMed PMID: 22102449.
28: Matus S, Glimcher LH, Hetz C. Protein folding stress in neurodegenerative diseases: a glimpse into the ER. Curr Opin Cell Biol. 2011 Apr;23(2):239-52. doi: 10.1016/j.ceb.2011.01.003. Epub 2011 Jan 31. Review. PubMed PMID: 21288706.
29: Nijholt DA, de Graaf TR, van Haastert ES, Oliveira AO, Berkers CR, Zwart R, Ovaa H, Baas F, Hoozemans JJ, Scheper W. Endoplasmic reticulum stress activates autophagy but not the proteasome in neuronal cells: implications for Alzheimer’s disease. Cell Death Differ. 2011 Jun;18(6):1071-81. doi: 10.1038/cdd.2010.176. Epub 2011 Jan 21. PubMed PMID: 21252911; PubMed Central PMCID: PMC3131935.
30: Dysken MW, Sano M, Asthana S, Vertrees JE, Pallaki M, Llorente M, Love S, Schellenberg GD, McCarten JR, Malphurs J, Prieto S, Chen P, Loreck DJ, Trapp G, Bakshi RS, Mintzer JE, Heidebrink JL, Vidal-Cardona A, Arroyo LM, Cruz AR, Zachariah S, Kowall NW, Chopra MP, Craft S, Thielke S, Turvey CL, Woodman C, Monnell KA, Gordon K, Tomaska J, Segal Y, Peduzzi PN, Guarino PD. Effect of Vitamin E and Memantine on Functional Decline in Alzheimer Disease: The TEAM-AD VA Cooperative Randomized Trial. JAMA. 2014 Jan 1;311(1):33-44. doi: 10.1001/jama.2013.282834. PubMed PMID: 24381967.
31: Moreno JA, Radford H, Peretti D, Steinert JR, Verity N, Martin MG, Halliday M, Morgan J, Dinsdale D, Ortori CA, Barrett DA, Tsaytler P, Bertolotti A, Willis AE, Bushell M, Mallucci GR. Sustained translational repression by eIF2α-P mediates prion neurodegeneration. Nature. 2012 May 6;485(7399):507-11. doi: 10.1038/nature11058. PubMed PMID: 22622579; PubMed Central PMCID: PMC3378208.
32: Shen S, Kepp O, Kroemer G. The end of autophagic cell death? Autophagy. 2012 Jan;8(1):1-3. doi: 10.4161/auto.8.1.16618. Epub 2012 Jan 1. Review. PubMed PMID: 22082964.
33: Gordy C, He YW. The crosstalk between autophagy and apoptosis: where does this lead? Protein Cell. 2012 Jan;3(1):17-27. doi: 10.1007/s13238-011-1127-x. Epub 2012 Feb 9. Review. PubMed PMID: 22314807.
34: Moreno JA, Halliday M, Molloy C, Radford H, Verity N, Axten JM, Ortori CA, Willis AE, Fischer PM, Barrett DA, Mallucci GR. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci Transl Med. 2013 Oct 9;5(206):206ra138. doi: 10.1126/scitranslmed.3006767. PubMed PMID: 24107777.
35: Axten JM, Medina JR, Feng Y, Shu A, Romeril SP, Grant SW, Li WH, Heerding DA, Minthorn E, Mencken T, Atkins C, Liu Q, Rabindran S, Kumar R, Hong X, Goetz A, Stanley T, Taylor JD, Sigethy SD, Tomberlin GH, Hassell AM, Kahler KM, Shewchuk LM, Gampe RT. Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1H-indol-5-yl)-7H-p yrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). J Med Chem. 2012 Aug 23;55(16):7193-207. doi: 10.1021/jm300713s. Epub 2012 Aug 8. PubMed PMID: 22827572.
36: Freed CR, Greene PE, Breeze RE, Tsai WY, DuMouchel W, Kao R, Dillon S, Winfield H, Culver S, Trojanowski JQ, Eidelberg D, Fahn S. Transplantation of embryonic dopamine neurons for severe Parkinson’s disease. N Engl J Med. 2001 Mar 8;344(10):710-9. PubMed PMID: 11236774.
37: Ma T, Trinh MA, Wexler AJ, Bourbon C, Gatti E, Pierre P, Cavener DR, Klann E. Suppression of eIF2α kinases alleviates Alzheimer’s disease-related plasticity and memory deficits. Nat Neurosci. 2013 Sep;16(9):1299-305. doi: 10.1038/nn.3486. Epub 2013 Aug 11. PubMed PMID: 23933749; PubMed Central PMCID: PMC3756900.
38: Janssens J, Van Broeckhoven C. Pathological mechanisms underlying TDP-43 driven neurodegeneration in FTLD-ALS spectrum disorders. Hum Mol Genet. 2013 Oct 15;22(R1):R77-87. doi: 10.1093/hmg/ddt349. Epub 2013 Jul 29. PubMed PMID: 23900071; PubMed Central PMCID: PMC3782069.
39: Kim HJ, Raphael AR, Ladow ES, McGurk L, Weber RA, Trojanowski JQ, Lee VM, Finkbeiner S, Gitler AD, Bonini NM. Therapeutic modulation of eIF2α phosphorylation rescues TDP-43 toxicity in amyotrophic lateral sclerosis disease models. Nat Genet. 2013 Dec 15. doi: 10.1038/ng.2853. [Epub ahead of print] PubMed PMID: 24336168.
40: Trinh MA, Kaphzan H, Wek RC, Pierre P, Cavener DR, Klann E. Brain-specific disruption of the eIF2α kinase PERK decreases ATF4 expression and impairs behavioral flexibility. Cell Rep. 2012 Jun 28;1(6):676-88. doi: 10.1016/j.celrep.2012.04.010. Epub 2012 May 24. PubMed PMID: 22813743; PubMed Central PMCID: PMC3401382.
41: Zhu PJ, Huang W, Kalikulov D, Yoo JW, Placzek AN, Stoica L, Zhou H, Bell JC, Friedlander MJ, Krnjević K, Noebels JL, Costa-Mattioli M. Suppression of PKR promotes network excitability and enhanced cognition by interferon-γ-mediated disinhibition. Cell. 2011 Dec 9;147(6):1384-96. doi: 10.1016/j.cell.2011.11.029. PubMed PMID: 22153080; PubMed Central PMCID: PMC3569515.
42: Costa-Mattioli M, Gobert D, Stern E, Gamache K, Colina R, Cuello C, Sossin W, Kaufman R, Pelletier J, Rosenblum K, Krnjević K, Lacaille JC, Nader K, Sonenberg N. eIF2alpha phosphorylation bidirectionally regulates the switch from short- to long-term synaptic plasticity and memory. Cell. 2007 Apr 6;129(1):195-206. PubMed PMID: 17418795.
43: Jiang Z, Belforte JE, Lu Y, Yabe Y, Pickel J, Smith CB, Je HS, Lu B, Nakazawa K. eIF2alpha Phosphorylation-dependent translation in CA1 pyramidal cells impairs hippocampal memory consolidation without affecting general translation. J Neurosci. 2010 Feb 17;30(7):2582-94. doi: 10.1523/JNEUROSCI.3971-09.2010. PubMed PMID: 20164343; PubMed Central PMCID: PMC2836228.
44: Sidrauski C, Acosta-Alvear D, Khoutorsky A, Vedantham P, Hearn BR, Li H, Gamache K, Gallagher CM, Ang KK, Wilson C, Okreglak V, Ashkenazi A, Hann B, Nader K, Arkin MR, Renslo AR, Sonenberg N, Walter P. Pharmacological brake-release of mRNA translation enhances cognitive memory. Elife. 2013 May 28;2:e00498. doi: 10.7554/eLife.00498. Print 2013. PubMed PMID: 23741617; PubMed Central PMCID: PMC3667625.
45: Gao Y, Sartori DJ, Li C, Yu QC, Kushner JA, Simon MC, Diehl JA. PERK is required in the adult pancreas and is essential for maintenance of glucose homeostasis. Mol Cell Biol. 2012 Dec;32(24):5129-39. doi: 10.1128/MCB.01009-12. Epub 2012 Oct 15. PubMed PMID: 23071091; PubMed Central PMCID: PMC3510549.
46: Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010 Mar 19;140(6):900-17. doi: 10.1016/j.cell.2010.02.034. Review. PubMed PMID: 20303879; PubMed Central PMCID: PMC2887297.
47: Marciniak SJ, Ron D. Endoplasmic reticulum stress signaling in disease. Physiol Rev. 2006 Oct;86(4):1133-49. Review. PubMed PMID: 17015486.
48: Scheuner D, Vander Mierde D, Song B, Flamez D, Creemers JW, Tsukamoto K, Ribick M, Schuit FC, Kaufman RJ. Control of mRNA translation preserves endoplasmic reticulum function in beta cells and maintains glucose homeostasis. Nat Med. 2005 Jul;11(7):757-64. Epub 2005 Jun 26. PubMed PMID: 15980866.
49: Cornejo VH, Pihán P, Vidal RL, Hetz C. Role of the unfolded protein response in organ physiology: Lessons from mouse models. IUBMB Life. 2013 Dec;65(12):962-75. doi: 10.1002/iub.1224. Epub 2013 Nov 13. PubMed PMID: 24227223.
50: Aviles-Olmos I, Dickson J, Kefalopoulou Z, Djamshidian A, Ell P, Soderlund T, Whitton P, Wyse R, Isaacs T, Lees A, Limousin P, Foltynie T. Exenatide and the treatment of patients with Parkinson’s disease. J Clin Invest. 2013 Jun 3;123(6):2730-6. PubMed PMID: 23728174; PubMed Central PMCID: PMC3668846.
51: Chen A, Muzzio IA, Malleret G, Bartsch D, Verbitsky M, Pavlidis P, Yonan AL, Vronskaya S, Grody MB, Cepeda I, Gilliam TC, Kandel ER. Inducible enhancement of memory storage and synaptic plasticity in transgenic mice expressing an inhibitor of ATF4 (CREB-2) and C/EBP proteins. Neuron. 2003 Aug 14;39(4):655-69. PubMed PMID: 12925279.
52: Gomperts SN, Locascio JJ, Rentz D, Santarlasci A, Marquie M, Johnson KA, Growdon JH. Amyloid is linked to cognitive decline in patients with Parkinson disease without dementia. Neurology. 2013 Jan 1;80(1):85-91. doi: 10.1212/WNL.0b013e31827b1a07. Epub 2012 Dec 12. PubMed PMID: 23243071; PubMed Central PMCID: PMC3589197.
53: Lim YY, Maruff P, Pietrzak RH, Ellis KA, Darby D, Ames D, Harrington K, Martins RN, Masters CL, Szoeke C, Savage G, Villemagne VL, Rowe CC; AIBL Research Group. Aβ and cognitive change: Examining the preclinical and prodromal stages of Alzheimer’s disease. Alzheimers Dement. 2014 Feb 28. pii: S1552-5260(13)02939-7. doi: 10.1016/j.jalz.2013.11.005. [Epub ahead of print] PubMed PMID: 24589436.
54: Lim YY, Maruff P, Pietrzak RH, Ames D, Ellis KA, Harrington K, Lautenschlager NT, Szoeke C, Martins RN, Masters CL, Villemagne VL, Rowe CC; AIBL Research Group. Effect of amyloid on memory and non-memory decline from preclinical to clinical Alzheimer’s disease. Brain. 2014 Jan;137(Pt 1):221-31. doi: 10.1093/brain/awt286. Epub 2013 Oct 30. PubMed PMID: 24176981.
55: Lim YY, Ellis KA, Pietrzak RH, Ames D, Darby D, Harrington K, Martins RN, Masters CL, Rowe C, Savage G, Szoeke C, Villemagne VL, Maruff P; AIBL Research Group. Stronger effect of amyloid load than APOE genotype on cognitive decline in healthy older adults. Neurology. 2012 Oct 16;79(16):1645-52. doi: 10.1212/WNL.0b013e31826e9ae6. PubMed PMID: 23071163.
56: Doraiswamy PM, Sperling RA, Coleman RE, Johnson KA, Reiman EM, Davis MD, Grundman M, Sabbagh MN, Sadowsky CH, Fleisher AS, Carpenter A, Clark CM, Joshi AD, Mintun MA, Skovronsky DM, Pontecorvo MJ; AV45-A11 Study Group. Amyloid-β assessed by florbetapir F 18 PETand 18-month cognitive decline: a multicenter study. Neurology. 2012 Oct 16;79(16):1636-44. doi: 10.1212/WNL.0b013e3182661f74. Epub 2012 Jul 11. PubMed PMID: 22786606; PubMed Central PMCID: PMC3468774.
57: Landau SM, Mintun MA, Joshi AD, Koeppe RA, Petersen RC, Aisen PS, Weiner MW, Jagust WJ; Alzheimer’s Disease Neuroimaging Initiative. Amyloid deposition, hypometabolism, and longitudinal cognitive decline. Ann Neurol. 2012 Oct;72(4):578-86. doi: 10.1002/ana.23650. PubMed PMID: 23109153; PubMed Central PMCID: PMC3786871.
58: Wilson RS, Leurgans SE, Boyle PA, Schneider JA, Bennett DA. Neurodegenerative basis of age-related cognitive decline. Neurology. 2010 Sep 21;75(12):1070-8. Epub 2010 Sep 15. PubMed PMID: 20844243; PubMed Central PMCID: PMC2942064
59: Cakir I, Cyr NE, Perello M, Litvinov BP, Romero A, Stuart RC, Nillni EA. Obesity induces hypothalamic endoplasmic reticulum stress and impairs proopiomelanocortin (POMC) post-translational processing. J Biol Chem. 2013 Jun 14;288(24):17675-88. doi: 10.1074/jbc.M113.475343. Epub 2013 May 2. PubMed PMID: 23640886; PubMed Central PMCID: PMC3682568.
60: Hosoi T, Sasaki M, Miyahara T, Hashimoto C, Matsuo S, Yoshii M, Ozawa K. Endoplasmic reticulum stress induces leptin resistance. Mol Pharmacol. 2008 Dec;74(6):1610-9. doi: 10.1124/mol.108.050070. Epub 2008 Aug 28. PubMed PMID: 18755873.
61: Ozcan L, Ergin AS, Lu A, Chung J, Sarkar S, Nie D, Myers MG Jr, Ozcan U. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab. 2009 Jan 7;9(1):35-51. doi: 10.1016/j.cmet.2008.12.004. PubMed PMID: 19117545.
62: Casas-Tinto S, Zhang Y, Sanchez-Garcia J, Gomez-Velazquez M, Rincon-Limas DE, Fernandez-Funez P. The ER stress factor XBP1s prevents amyloid-beta neurotoxicity. Hum Mol Genet. 2011 Jun 1;20(11):2144-60. doi: 10.1093/hmg/ddr100. Epub 2011 Mar 9. PubMed PMID: 21389082; PubMed Central PMCID: PMC3090193.
63: Liu SY, Wang W, Cai ZY, Yao LF, Chen ZW, Wang CY, Zhao B, Li KS. Polymorphism -116C/G of human X-box-binding protein 1 promoter is associated with risk of Alzheimer’s disease. CNS Neurosci Ther. 2013 Apr;19(4):229-34. doi: 10.1111/cns.12064. Epub 2013 Feb 20. PubMed PMID: 23421912.
64: Teske BF, Wek SA, Bunpo P, Cundiff JK, McClintick JN, Anthony TG, Wek RC. The eIF2 kinase PERK and the integrated stress response facilitate activation of ATF6 during endoplasmic reticulum stress. Mol Biol Cell. 2011 Nov;22(22):4390-405. doi: 10.1091/mbc.E11-06-0510. Epub 2011 Sep 14. PubMed PMID: 21917591; PubMed Central PMCID: PMC3216664.
65: Loewen CA, Feany MB. The unfolded protein response protects from tau neurotoxicity in vivo. PLoS One. 2010 Sep 29;5(9). pii: e13084. doi: 10.1371/journal.pone.0013084. PubMed PMID: 20927324; PubMed Central PMCID: PMC2947516
66: Unterberger U, Höftberger R, Gelpi E, Flicker H, Budka H, Voigtländer T. Endoplasmic reticulum stress features are prominent in Alzheimer disease but not in prion diseases in vivo. J Neuropathol Exp Neurol. 2006 Apr;65(4):348-57. PubMed PMID: 16691116.
67: Höglinger GU, Melhem NM, Dickson DW, Sleiman PM, Wang LS, Klei L, Rademakers R, de Silva R, Litvan I, Riley DE, van Swieten JC, Heutink P, Wszolek ZK, Uitti RJ, Vandrovcova J, Hurtig HI, Gross RG, Maetzler W, Goldwurm S, Tolosa E, Borroni B, Pastor P; PSP Genetics Study Group, Cantwell LB, Han MR, Dillman A, van der Brug MP, Gibbs JR, Cookson MR, Hernandez DG, Singleton AB, Farrer MJ, Yu CE, Golbe LI, Revesz T, Hardy J, Lees AJ, Devlin B, Hakonarson H, MüllerU, Schellenberg GD. Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat Genet. 2011 Jun 19;43(7):699-705. doi: 10.1038/ng.859. PubMed PMID: 21685912; PubMed Central PMCID: PMC3125476.
68: Egawa N, Yamamoto K, Inoue H, Hikawa R, Nishi K, Mori K, Takahashi R. The endoplasmic reticulum stress sensor, ATF6α, protects against neurotoxin-induced dopaminergic neuronal death. J Biol Chem. 2011 Mar 11;286(10):7947-57. doi: 10.1074/jbc.M110.156430. Epub 2010 Dec 3. PubMed PMID: 21131360; PubMed Central PMCID: PMC3048681.
69: Valdés P, Mercado G, Vidal RL, Molina C, Parsons G, Court FA, Martinez A, Galleguillos D, Armentano D, Schneider BL, Hetz C. Control of dopaminergic neuron survival by the unfolded protein response transcription factor XBP1. Proc Natl Acad Sci U S A. 2014 May 6;111(18):6804-9. doi: 10.1073/pnas.1321845111. Epub 2014 Apr 21. PubMed PMID: 24753614; PubMed Central PMCID: PMC4020088.
70: Fouillet A, Levet C, Virgone A, Robin M, Dourlen P, Rieusset J, Belaidi E, Ovize M, Touret M, Nataf S, Mollereau B. ER stress inhibits neuronal death by promoting autophagy. Autophagy. 2012 Jun;8(6):915-26. doi: 10.4161/auto.19716. Epub 2012 Jun 1. PubMed PMID: 22660271; PubMed Central PMCID: PMC3427257.
71: Yamamoto K, Sato T, Matsui T, Sato M, Okada T, Yoshida H, Harada A, Mori K. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev Cell. 2007 Sep;13(3):365-76. PubMed PMID: 17765680.
72: Walker LC, Diamond MI, Duff KE, Hyman BT. Mechanisms of protein seeding in neurodegenerative diseases. JAMA Neurol. 2013 Mar 1;70(3):304-10. Review. PubMed PMID: 23599928; PubMed Central PMCID: PMC3665718.
73: Strobel G. In Revival of Parabiosis, Young Blood Rejuvenates Aging Microglia, Cognition. Alzforum. 2014 May 05. Online Resource. Available at http://www.alzforum.org/news/conference-coverage/revival-parabiosis-young-blood-rejuvenates-aging-microglia-cognition . Accessed 2014-09-20. Summarizing presentation by Anton Wyss-Coray at the Zilkha Symposium on Alzheimer’s Disease and Related Disorders, Keck School of Medicine, University of Southern California, Los Angeles, 2014 April 4.
74: Katsimpardi L, Litterman NK, Schein PA, Miller CM, Loffredo FS, Wojtkiewicz GR, Chen JW, Lee RT, Wagers AJ, Rubin LL. Vascular and neurogenic rejuvenation of the aging mouse brain by young systemic factors. Science. 2014 May 9;344(6184):630-4. doi: 10.1126/science.1251141. Epub 2014 May 5. PubMed PMID: 24797482; PubMed Central PMCID: PMC4123747.
75: Villeda SA, Plambeck KE, Middeldorp J, Castellano JM, Mosher KI, Luo J, Smith LK, Bieri G, Lin K, Berdnik D, Wabl R, Udeochu J, Wheatley EG, Zou B, Simmons DA, Xie XS, Longo FM, Wyss-Coray T. Young blood reverses age-related impairments in cognitive function and synaptic plasticity in mice. Nat Med. 2014 Jun;20(6):659-63. doi: 10.1038/nm.3569. Epub 2014 May 4. PubMed PMID: 24793238.
76: Ruckh JM, Zhao JW, Shadrach JL, van Wijngaarden P, Rao TN, Wagers AJ, Franklin RJ. Rejuvenation of regeneration in the aging central nervous system. Cell Stem Cell. 2012 Jan 6;10(1):96-103. doi: 10.1016/j.stem.2011.11.019. PubMed PMID: 22226359; PubMed Central PMCID: PMC3714794.
77: Villeda SA, Luo J, Mosher KI, Zou B, Britschgi M, Bieri G, Stan TM, Fainberg N, Ding Z, Eggel A, Lucin KM, Czirr E, Park JS, Couillard-Després S, Aigner L, Li G, Peskind ER, Kaye JA, Quinn JF, Galasko DR, Xie XS, Rando TA, Wyss-Coray T. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature. 2011 Aug 31;477(7362):90-4. doi: 10.1038/nature10357. PubMed PMID: 21886162; PubMed Central PMCID: PMC3170097.
78: Lee SR, Lee HJ, Cha SH, Jeong KJ, Lee YJ, Jeon CY, Yi KS, Lim I, Chang ZH, Kim SU. Long-term survival and differentiation ofhuman neural stem cells in nonhuman primate brain with no immunosuppression. Cell Transplant. 2014 Jan 29. doi: 10.3727/096368914X678526. [Epub ahead of print] PubMed PMID: 24480401.
79: Hemmer K, Zhang M, van Wüllen T, Sakalem M, Tapia N, Baumuratov A, Kaltschmidt C, Kaltschmidt B, Schöler HR, Zhang W, Schwamborn JC. Induced neural stem cells achieve long-term survival and functional integration in the adult mouse brain. Stem Cell Reports. 2014 Sep 9;3(3):423-31. doi: 10.1016/j.stemcr.2014.06.017. Epub 2014 Jul 31. PubMed PMID: 25241741.
80: Rae MJ, Butler RN, Campisi J, de Grey AD, Finch CE, Gough M, Martin GM, Vijg J, Perrott KM, Logan BJ. The demographic and biomedical case for late-life interventions in aging. Sci Transl Med. 2010 Jul 14;2(40):40cm21. doi: 10.1126/scitranslmed.3000822. PubMed PMID: 20630854; PubMed Central PMCID: PMC3677970.