
Immunotherapy targeting the age-related accumulation of extracellular aggregates, in the form of ß-amyloid, is the first rejuvenation biotechnology to reach Phase III human clinical trials. The promise of this therapy for the treatment and prevention of Alzheimer’s disease (and ultimately, of so-called “normal” brain aging) has sparked an interest in utilizing the same approach for other forms of aging damage, including the clearance of aggregated intracellular proteinaceous aging damage. Notably, as we have reviewed in a series of four previous posts, recent years have seen the appearance of a rising number preclinical studies of therapeutic vaccines targeting pathological tau species accumulating in the brains and spinal cords of transgenic rodent models of tauopathic neurodegeneration. These studies have reported — somewhat surprisingly — the antibody-mediated clearance of these primarily intracellular aging lesions, accompanied by functional improvements in treated animals.(4-8) These two forms of structural damage are major contributors to the age-related degeneration of the brain, whether it leads to frank dementia or to the diagnostic euphemism of “normal” age-related cognitive decline, and novel therapeutics to effect the removal of both from aging neurons will be key elements of a comprehensive panel of rejuvenation biotechnologies.
One limitation to the existing studies of vaccines targeting pathological tau has been that they have tended to initiate therapy before, at, or only very shortly after the appearance of tau pathology in the murine rodent system, and of the onset of cognitive or motor deficits — a luxury which we cannot expect to be afforded in treating cases of frank dementia related to these inclusions, and that may not reflect the condition of the brain even prior to the appearance of gross cognitive and functional deficits in aging humans. Further complicating these matters, the combination of early intervention and the very rapid progression of tau pathology in these models has meant that these studies have given only limited information on the effects of the onset or later clearance of neurofibrillary tangles (NFT), which are the most prominent and mature form of tau pathology to appear in the brain during aging, in Alzheimer’s disease, and with the genetic tauopathies. A group headed by Dr. Lars Ittner of the Laboratory for Translational Neurodegeneration at the University of Sydney, Camperdown has now provided the first evidence on these unresolved questions, furthering the case for human therapies targeting pathological tau.
Ittner’s group performed their studies in pR5 mice, which express the mutant human tau species P301L, which is responsible for the human dementing tauopathy frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17). These mice begin to accumulate tau that has been hyperphosphorylated at different epitopes early in life, but phosphorylation of the NFT-associated Ser422 and Ser396/S404 (the paired helical fragment-1 (PHF-1) epitope) first begins to appear at the 6 mo mark, accompanied by NFT pathology, which progresses thereafter throughout their shortened lifespans. The investigators accordingly engineered a vaccine targeting these NFT-associated species by linking a human tau peptide comprising phosphorylated PHF-1 Ser396/Ser404 to the immunogenic carrier protein KLH. This vaccine was then administered to animals before (4 mo), shortly after (8 mo), and long after (18 mo) the 6-month age of NFT onset, accompanied by complete Freund’s adjuvant, and later followed by further adjuvant booster.(1)
By 17 mo, hippocampal and amygdalic neurons of untreated mice exhibited high levels of antibody staining for phorphoylated NFT-associated tau epitopes; even more notably, staining was also high in the dystrophic neurites in subregions of these areas. Immunization targeting NFT tau epitopes had no effect on expression or distribution of either human mutant or total (transgenic plus native murine native) tau, but did result in robust (≥1:1000) antigen titers within four months of vaccination. Notably, even untreated pR5 mice exhibited low levels of such titers, while no such antibodies were evident in isochronic wild-type mice; this suggests a (weak) intrinsic immune response against the presence of aberrant tau species in mice bearing them.(1) The existence of such a response is consistent with previous findings in aging humans,(3) Alzheimer’s disease patients,(3) and transgenic murine tauopathy.(2)
Tau-targeting vaccination substantially reduced the subsequent burden of pathological tau in neurons and dystrophic neurites, no matter when treatment was initiated:
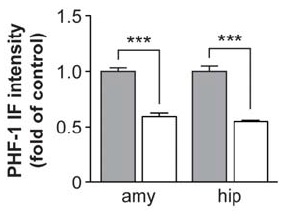
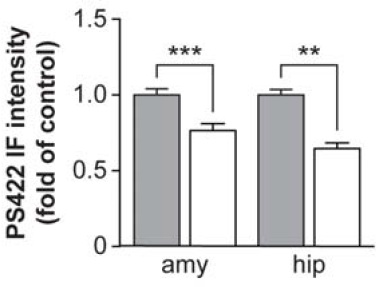
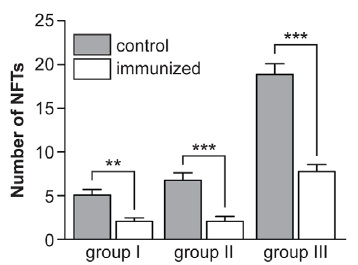
Figure 1. Vaccination targeting pathological tau species reduces the further spread of antibody-labeled PHF-1 (left) and tau phosphorylated at Ser422 (center) when initiated shortly after NFT appearance; when administered before (Group I), shortly after (Group II), or long after (Group III) their appearance, it similarly impeded the appearance of Gallyas silver-stained NFTs per se (right). Reproduced from (1) under the terms of Creative Commons License.
Limitations, and their Supercedence
One key limitation to this study is that no cognitive or motor function data are presented. In prior studies, however, preventive and therapeutic vaccination in similar murine models of tau neuropathy has led to relative improvements in such functions, proportional to the reduction in neuropathology.
Another limitation is of interpretation of the results, and possibly of the actual effects of the vaccine therapy itself. The authors note that the burden of NFT in treated animals from Group III — aged 22 mo at autopsy, but whose immunization was delayed until 18 mo of age (ie, a full year after the onset of neuropathology) — was similar to that in untreated mice shortly before (Group I) or shortly after (Group II) that onset at autopsy. Because autopsied Group II animals were of similar age to the the age at which vaccination was initiated in Group III, and unvaccinated Group II animals’ neuropathology burden was similar to that present months after treatment in animals that were initially treated at a similar age, the authors suggest that the apparently similar NFT burdens after several months of vaccination in the latter group suggest that the treatment had arrested the further accumulation of tau pathology following vaccination, rather than having actively cleared it. However, as they also note, it is also possible that their tau vaccine did remove existing tau inclusions, but that the rate of active clearance of tau pathology was equal to the rate of new NFT accumulation.(1) This is quite plausible, granted the very rapid progression of tau pathology in these animals when left untreated, and even more so granted their relatively brief period of treatment: Group III was sufficiently far into their shortened life expectancy as to necessitate abbreviated (6 mo) therapy, relative to the 9-10 mo afforded to Groups I and II. Thus, a longer period of antibody-mediated clearance might have afforded a more thorough therapeutic effect.
At a minimum, even a vaccine that only arrested the burden of tau pathology could contribute to the prevention of significant age-related cognitive decline if administered in middle age. And in aging humans, the development of neurofibrillary tangles occurs over the course of decades, rather than months as in these animals; thus, even a moderate rate of true clearance could in principle be sufficient to lead to a net reduction in NFT burden in humans, and thus to the reversal of one component of brain aging.
In either case, several strategies could be explored to enhance the efficacy of such a vaccine. The most obvious is the administration of a series of booster shots, to increase therapeutic antibody titers or to restore putatively flagging numbers in the months (or in humans, perhaps years) after initial vaccination. Note that the investigators only collected a single data point on antibodies targeting pathological tau, taken relatively soon (4 mo) after a single vaccination at a single antigen dose, following which the same, single round of vaccination was left to work for a further 6 (Groups 1 and II) or 2 (Group III) months in younger and older animals, respectively; this clearly leaves open the potential that a series of rounds of treatment could have been more effective.
Another potential way to enhance the efficacy of a similar vaccine in humans would be to fortify the lysosome with novel hydrolytic enzymes, to enhance its capacity to clear out aggregates which might be targeted to the lysosome by anti-phospho-tau antibodies. While the involvement of lysosomal degradation of aberrant tau species in the therapeutic effects of immunotherapy was not evaluated in this study,(1) such an effect is consistent with the clearance of intraneuronal phospho-tau in these vaccination studies, and with much other data. Vaccination of a Parkinson’s disease transgenic mouse model with α-synuclein, similarly, leads to a reduction of intracellular α-synuclein aggregates;(11) several studies have found that immunotherapy targeting β-amyloid leads to a reduction in intraneuronal forms of the aggregate, and work in Gunnar Gouras’ laboratory, reviewed in an earlier post, finds that antibodies against Aβ can be internalized in AD neuronal culture models of Aβ accumulation and clear intraneuronal Aβ aggregates via the endosomal–lysosomal pathway. Additionally, the authors of the current study(1) also note that immunized K257T/P301S tauopathy mice exhibit changes in cathepsin levels, consistent with lysosomal degradation of pathological tau;(6) in this context, it is notable that the lysosomal-enhancing drug PADK (see a previous post) removes PHF-1 from hippocampal slices, in association with a normalization of impaired synaptic integrity.(10,11) I will note that the authors of the current study(1) do argue against such a mechanism, on the basis that “total tau levels remained unchanged upon immunization of JNPL3 mice [(5) below], and staining with total tau antibodies was comparable in immunized and control pR5 mice (this study), arguing against overtly increased degradation of tau” — but of course, this does not argue against specific degradation of pathological tau species targeted by immunotherapy in their study.(1)
An additional possibility is that the clearance of other intraneuronal aggregates from aging neurons may itself facilitate the additional proteolytic or lysosomal removal of phospho-tau species. The relationship between the accumulation and neurotoxicity of Aβ and aberrant tau is complex,(13) but reducing one will almost certainly reduce the deleterious effects of the other; and, in particular, immunization against Aβ has been reported to lead to the clearance of early phospho-tau species.(14 (and see (15)) This adds a further argument in favor of a comprehensive approach to brain aging and Alzheimer’s disease, suggesting synergistic rather than merely additive effects of combining therapies targeting Aβ in addition to malformed tau.
Finally, it is also possible that future vaccines might be more specifically targeted to especially pathologically important tau species, or be designed to elicit antibodies with higher avidity for their target. All of these avenues deserve exploration in future studies of this promising therapeutic approach.
Breaking from a March to a Run
This latest report(1) adds to a series of previous successful preclinical studies(4-8) of immunotherapies targeting pathological tau species for therapeutic clearance. This new study is notable for marking the entry of a new group of investigators into the tau vaccine space, suggesting that this area of rejuvenation biotechnology is attracting an increasing number of independent research teams. While limited in scope and in design, the new study adds to prior knowledge not only by testing a new vaccine, but importantly by evaluating its effects in animals in whom damage from tau inclusions was already extensive. While cognitive and motor functions were not evaluated, the clear ability of this vaccine to arrest — and perhaps to reverse — the burden of a life-long, accelerated deposition of aggressive pathological tau species offers promise for human translation into a treatment for genetic tauopathies, Alzheimer’s disease, and the “normal” age-related decay of cogntitive, emotional, and neurological function that is only now finally being recognized as a disorder meriting therapy — and ultimately, cure. The gaps in this study are opportunities for further experimentation, further refinement, further learning, and further therapeutic development; it marks progress, bringing us closer to the goal of a comprehensive panel of rejuvenation biotechnologies and the rescue of aging bodies and minds.
References
1: Bi M, Ittner A, Ke YD, Götz J, Ittner LM. Tau-Targeted Immunization Impedes Progression of Neurofibrillary Histopathology in Aged P301L Tau Transgenic Mice. PLoS One. 2011;6(12):e26860. Epub 2011 Dec 8. PubMed PMID: 22174735; PubMed Central PMCID: PMC3234245.
2: Rosenmann H, Grigoriadis N, Karussis D, Boimel M, Touloumi O, Ovadia H, Abramsky O. Tauopathy-like abnormalities and neurologic deficits in mice immunized with neuronal tau protein. Arch Neurol. 2006 Oct;63(10):1459-67. PubMed PMID: 17030663.
3: Rosenmann H, Meiner Z, Geylis V, Abramsky O, Steinitz M. Detection of circulating antibodies against tau protein in its unphosphorylated and in its neurofibrillary tangles-related phosphorylated state in Alzheimer’s disease and healthy subjects. Neurosci Lett. 2006 Dec 20;410(2):90-3. PubMed PMID: 17095156.
4: Rosenmann H, Grigoriadis N, Karussis D, Boimel M, Touloumi O, Ovadia H, Abramsky O. Tauopathy-like abnormalities and neurologic deficits in mice immunized with neuronal tau protein. Arch Neurol. 2006 Oct;63(10):1459-67. PubMed PMID: 17030663.
5: Asuni AA, Boutajangout A, Quartermain D, Sigurdsson EM. Immunotherapy targeting pathological tau conformers in a tangle mouse model reduces brain pathology with associated functional improvements. J Neurosci. 2007 Aug 22;27(34):9115-29. PMID: 17715348 [PubMed – in process]
6: Boimel M, Grigoriadis N, Lourbopoulos A, Haber E, Abramsky O, Rosenmann H. Efficacy and safety of immunization with phosphorylated tau against neurofibrillary tangles in mice. Exp Neurol. 2010 Aug;224(2):472-85. Epub 2010 May 28. PubMed PMID: 20546729.
7: Boutajangout A, Ingadottir J, Davies P, Sigurdsson EM. Passive immunization targeting pathological phospho-tau protein in a mouse model reduces functional decline and clears tau aggregates from the brain. J Neurochem. 2011 Aug;118(4):658-67. doi: 10.1111/j.1471-4159.2011.07337.x. Epub 2011 Jul 1. PubMed PMID: 21644996.
8: Boutajangout A, Quartermain D, Sigurdsson EM. Immunotherapy targeting pathological tau prevents cognitive decline in a new tangle mouse model. J Neurosci. 2010 Dec 8;30(49):16559-66. PubMed PMID: 21147995.
9: Wisniewski T, Sigurdsson EM. Murine models of Alzheimer’s disease and their use in developing immunotherapies. Biochim Biophys Acta. 2010 Oct;1802(10):847-59. Epub 2010 May 13. PubMed PMID: 20471477; PubMed Central PMCID: PMC2930136.
10: Butler D, Brown QB, Chin DJ, Batey L, Karim S, Mutneja MS, Karanian DA, Bahr BA. Cellular responses to protein accumulation involve autophagy and lysosomal enzyme activation. Rejuvenation Res. 2005 Winter;8(4):227-37. PubMed PMID: 16313222.
11: Bendiske J, Bahr BA. Lysosomal activation is a compensatory response against protein accumulation and associated synaptopathogenesis–an approach for slowing Alzheimer disease? J Neuropathol Exp Neurol. 2003 May;62(5):451-63. PubMed PMID: 12769185.
12: Masliah E, Rockenstein E, Adame A, Alford M, Crews L, Hashimoto M, Seubert P, Lee M, Goldstein J, Chilcote T, Games D, Schenk D. Effects of alpha-synuclein immunization in a mouse model of Parkinson’s disease. Neuron. 2005 Jun 16;46(6):857-68. PMID: 15953415 [PubMed – indexed for MEDLINE]
13: Ittner LM, Götz J. Amyloid-β and tau–a toxic pas de deux in Alzheimer’s disease. Nat Rev Neurosci. 2011 Feb;12(2):65-72. Epub 2010 Dec 31. Review. PubMed PMID: 21193853.
14: Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM. Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron. 2004 Aug 5;43(3):321-32. PMID: 15294141 [PubMed – indexed for MEDLINE]
15: Oddo S, Caccamo A, Tran L, Lambert MP, Glabe CG, Klein WL, LaFerla FM. Temporal profile of amyloid-beta (Abeta) oligomerization in an in vivo model of Alzheimer disease. A link between Abeta and tau pathology. J Biol Chem. 2006 Jan 20;281(3):1599-604. Epub 2005 Nov 10. PMID: 16282321 [PubMed – indexed for MEDLINE]