Hi, my name is Shil Patel, and I graduated summa cum laude from the University of Pittsburgh in December 2016 with a BS in neuroscience and minors in chemistry and economics. Under the mentorship of Dr. Sharyl Fyffe-Maricich, my honor’s thesis examined the potential role of aberrant oligodendrocyte development as a leading cause of the symptoms in a neurodegenerative disorder called Rett Syndrome. Shortly after starting to work on this thesis during my junior year, I took a class on the development of the nervous system. With this brief introduction into the world of stem cells, I became fascinated with the potential of harnessing developmental pathways and processes for regenerative medicine. The following summer, I was awarded the Conte Center Summer Undergraduate Research Fellowship at the University of Pennsylvania School of Medicine where I got my first exposure to the world of somatic trans-differentiation, or, in other words, inducing adult cells to become different types of cells. This experience in the Hahn/Borgmann-Winter laboratory began to fuel my fascination with the role signaling cascades have in altering cellular function and identity.
Upon returning to the University of Pittsburgh, I joined Stephen Badylak’s laboratory. I worked on a project investigating the impact of extracellular matrix (ECM) hydrogels on esophageal cancer cell phenotype and function. Patients with esophageal adenocarcinoma (EAC) face a 16% 5-year survival rate, and esophagectomies, which are the current treatment standard, come with a 33% mortality rate that reflects the extreme nature of the procedure (Rubenstein & Shaheen, 2015). In collaboration with Dr. Blair Jobe, Dr. Badylak published a study in 2011 in which 5 patients with EAC had undergone resection of the esophageal mucosa and implantation of urinary bladder matrix (UBM) scaffolds over the denuded esophageal muscular layer. The scaffolds were degraded in the esophagus, releasing bio-active signaling molecules that induced an anti-inflammatory, reconstructive environment. After 13 months, normal and mature tissue was regenerated, replacing the resected tissue (Badylak et al., 2011). Currently, this work is in a Phase 1 clinical trial with the number of patients treated in the double digits, all of which exhibit no re-occurrence of the disease.
With these results, the Badylak laboratory has developed a method of enzymatically digesting the ECM scaffolds to release the resident bio-active materials, such as signaling molecules. This digested product forms a hydrogel and provides the opportunity to both study the bio-active components and explore the therapeutic potential of ECM hydrogels themselves. To this end, we investigated the effect of ECM hydrogels from two tissue sources on healthy, EAC precursor, and EAC cell lines in-vitro. Hydrogels derived from two different tissue types were tested and will be referred to generically as “hydrogel A” and “hydrogel B.” To determine their effect on metabolic activity, I used an assay that measures the quantity of the metabolic by-product NAD(P)H. I found that hydrogel A consistently decreased metabolic activity of only EAC cells. To assess cell proliferation, I used an assay that quantifies newly synthesized DNA. We found that treatment with hydrogel A decreases proliferation in one EAC cell type, while hydrogel B decreased proliferation in all cell types. The process of programmed or controlled cell death, called apoptosis, produces changes in the cell membrane. These changes can be marked by fluorescent staining, and a process called flow cytometry can count the stained cells. I stained the cells and a dedicated flow cytometry staff member performed the counts. While not statistically significant, my preliminary data suggests that treatment with either hydrogel causes increased death only in the EAC cell lines. Lastly, we were interested in seeing which gene networks may be causing these functional changes. After isolating RNA, the university Genomics Research Core ran a microarray to measure gene expression. We found hydrogel A differentially inhibited expression of certain genes in cancer cells while activating their expression in healthy cells. Further work will parse the transcriptome data to find the molecular networks most strongly involved in the functional changes in EAC cells that we have observed. This project demonstrates the promise of ECM degradation products as potential cancer therapeutics through their ability not only to selectively decrease cancerous functional characteristics, such as metabolic activity and proliferation, but also to selectively initiate programmed cell death in EAC cancer cells.
My interest in the field has continuously grown as I learn more about both the potential applications and the hurdles that have yet to be cleared. With regenerative medicine requiring contributions from many different fields, the SRF Summer Scholar project in the Loring laboratory is an incredible opportunity to become familiar with the process of using bioinformatics to approach the wealth of genetic data that may hold crucial insights into pathology and potential therapy.
Assessment of Genomic Integrity in Patient-Derived Fibroblasts, iPSCs and Neurons for Cell Therapy
This summer, I have joined Jeanne Loring’s laboratory at The Scripps Research Institute to explore stem cell treatment for Parkinson’s disease (PD). Parkinson’s affects between 7 and 10 million people worldwide. PD is characterized by a loss of neurons that produce and release the neurotransmitter dopamine, and this loss of “dopaminergic neurons” causes patients to experience progressive cognitive and motor deficits. In the 1970s, researchers began to explore the potential of replacing the lost neurons with new ones from fetal tissue. Despite mixed results in these early clinical trials, some patients experienced long-lasting improvement of PD symptoms and provided the proof of concept for further exploration of cell-based therapy (Bratt-Leal & Loring, 2016).
Due to the disadvantages of relying upon fetal tissue, the field has developed methods of generating dopaminergic neurons from a patient’s own skin cells (Kriks et al., 2011). With a virus that does not change the DNA of the skin cells or become pathogenic, we can activate four genes in adult cells that re-program them back into stem cells. These re-programmed cells are called induced pluripotent stem cells, or iPSCs, and they can be pushed to become dopaminergic neurons using combinations of different signaling molecules (Chambers et al., 2009). Studies in mice, rats and monkeys have shown that transplantation of these cells into models of PD can reverse symptoms, paving the way for clinical application (Kriks et al., 2011). As we move closer to clinical trials transplanting dopaminergic neurons from iPSCs, the importance of quality control rises greatly. The reprogramming process takes a single skin cell and converts it into a rapidly dividing pluripotent stem cell. We can analyze the DNA from those stem cells to ensure there aren’t dangerous duplications or deletions of genes. These duplications or deletions of genes are called copy number variations and may have been present in the original patient skin cells or been induced through prolonged time in cell culture. For example, if a gene that protects oncogenesis is deleted, transplanted neurons derived from those stem cells may form tumors in patients.

Figure 1. Generating Patient-Derived Dopaminergic Neurons.
Fibroblasts 10 patients will be re-programmed into induced pluripotent stem cells (iPSCs). To force iPSCs to become dopaminergic neurons, they will be subjected to differentiation and maturation media comprised of specific signaling molecules and growth factors for a 25-day period. The neurons can then be transplanted into the patient.
Thus, under the mentorship of Dr. Andres Bratt-Leal, I will be assessing the genotypes of induced pluripotent stem cell lines from 10 patients with PD to evaluate their genomic integrity. Single nucleotide polymorphisms (SNPs) are common sites of genetic variation between humans. By determining the precise nucleotide at common sites of variation, we begin to get a picture of the genome, and the more SNP sites we examine, the more the resolution of the genomic picture increases. I will load patient skin cell and iPSC DNA onto a microarray chip that can detect the nucleotide identity at 4.3 million SNPs across the genome and use bioinformatics software to identify CNVs. The cells will be compared against a reference human genome to see if harmful copy number variations were originally present in the skin cells or arose over their time in-vitro.
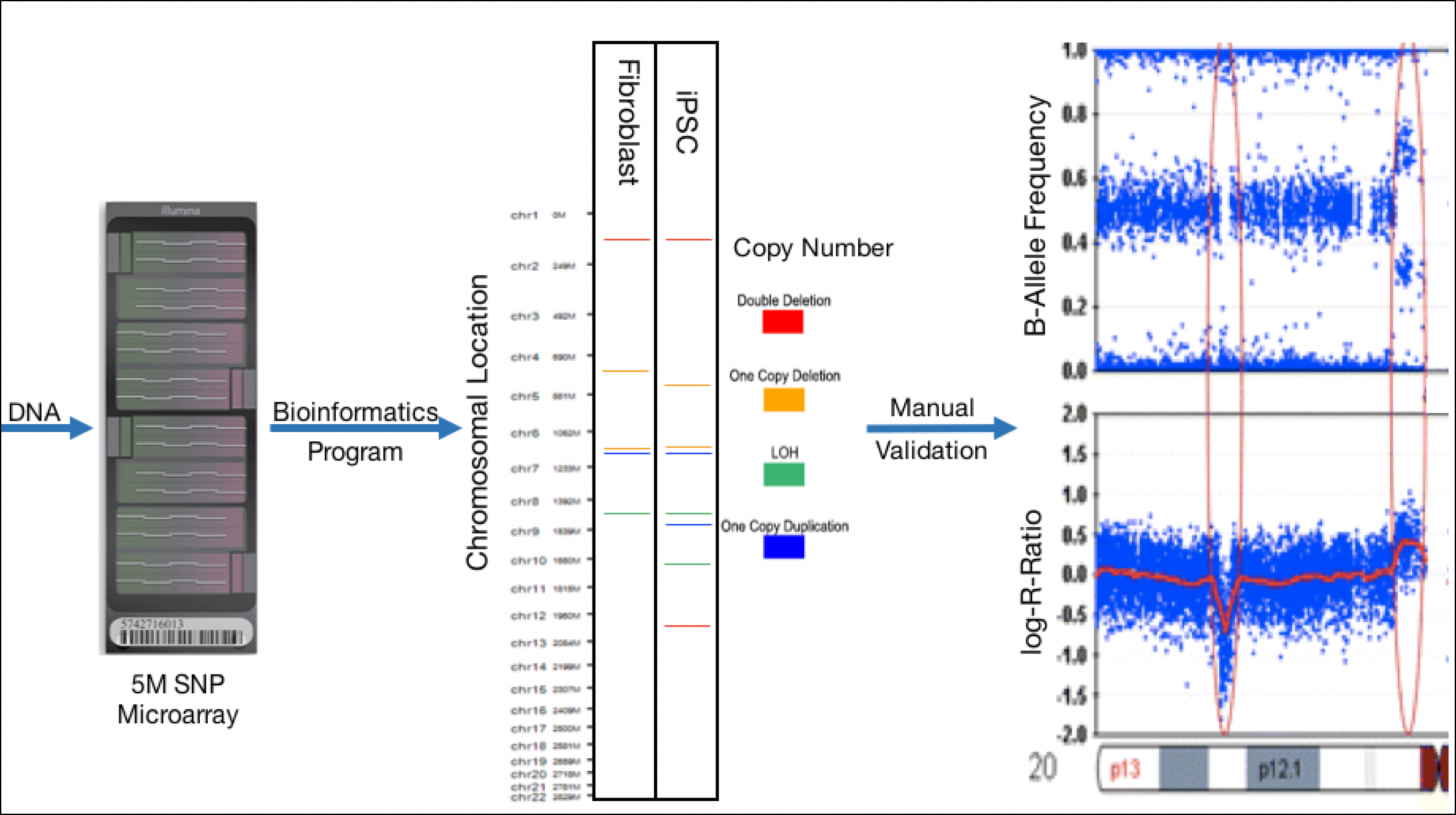
Figure 2. Workflow for CNV Discovery and Identification.
DNA will be isolated from fibroblasts and their resultant iPSC colonies and will be run on an Infinium Omni 5-4 microarray (Illumina). Copy number variations (CNVs) will be detected using bioinformatics programs with example results shown in step 2. Reported CNVs from the program will be manually verified by analysis of B-Allele Frequency and Log-R ratio plots. The above example plots represent a deletion in the first red ellipse and a duplication in the second.
The bioinformatics program CNVPartition will be used to preliminarily identify CNVs. Calculations called the “Log-R Ratio” and “B-Allele Frequency” each reveal different views of the SNP data and can be performed at each SNP site. Analyzed together, plots of these two calculations identify CNV presence and type. In order to verify or remove CNVs called by the CNVPartition program, I will manually evaluate the two plots, as shown in figure 2. Lastly, I will identify the genes that are affected by CNVs to determine if they may lead to disease.
The results will indicate which of the stem cell lines from each patient are safe to use for transplanting neurons. Using SNP microarrays to assess genomic integrity represents a high throughput quality control testing that can be used to safely create functional neurons from the cells of patients that require no immunosuppression after transplantation.
Future Plans:
After the SRF Summer Scholars Program, I will be joining the Rosandra Kaplan laboratory at the NIH’s National Cancer Institute. Under the post-baccalaureate Cancer Research Training Award, I will be studying the signaling processes that define tumor microenvironments and their impact on cellular function. During my second year at the NIH, I plan to apply to MD-PhD training programs.
References:
Badylak, S. F., Hoppo, T., Nieponice, A., Gilbert, T. W., Davison, J. M., & Jobe, B. A. (2011). Esophageal preservation in five male patients after endoscopic inner-layer circumferential resection in the setting of superficial cancer: a regenerative medicine approach with a biologic scaffold. Tissue Engineering Part A, 17(11-12), 1643-1650.
Bratt-Leal, A. M., & Loring, J. F. (2016). Stem Cells for Parkinson’s Disease Translational Neuroscience (pp. 187-201): Springer.
Chambers, S. M., Fasano, C. A., Papapetrou, E. P., Tomishima, M., Sadelain, M., & Studer, L. (2009). Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nature biotechnology, 27(3), 275-280.
Kriks, S., Shim, J.-W., Piao, J., Ganat, Y. M., Wakeman, D. R., Xie, Z., . . . Buch, A. (2011). Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson/’s disease. Nature, 480(7378), 547-551.
Rubenstein, J. H., & Shaheen, N. J. (2015). Epidemiology, diagnosis, and management of esophageal adenocarcinoma. Gastroenterology, 149(2), 302-317. e301.