MitoSENS
Preventing Damage from Mitochondrial Mutations
Mitochondria are the living machines within cells that act as their “power plants,” converting the energy-rich nutrients in our food into ATP, the form of energy that directly powers biochemical reactions in the cell. High-energy electrons are stripped from nutrients and used to fuel the four Complexes of the mitochondrial electron transport chain (ETS), which use the electrons’ energy to pump hydrogen ions across a membrane inside the organelle. Forcing so many charged ions onto one side of the membrane creates a reservoir of electrochemical potential energy, similar to the reservoir of potential energy created when water is held behind a dam in a hydroelectric generator. And just like a hydroelectric generator, that potential energy is channeled through a literal turbine (Complex V of the ETS), which stores the energy rushing through it as ATP.
Unlike any other part of the cell, mitochondria have their own mitochondrial DNA (mtDNA), which encodes 13 of the thousands of protein subunits of the ETS. But the majority of ETS proteins are encoded in the cell’s nucleus, as are all the proteins, organelles, and other structures made elsewhere in our cells. The reason that mitochondria uniquely have these genes is probably that they were entirely separate organisms at one point in our evolutionary history, with which our ancient single-celled ancestors developed a symbiotic relationship.
But just like real power plants, mitochondria generate toxic waste products in the process of “burning” food energy as fuel – in this case, spewing out highly-reactive molecules called free radicals, which can damage cellular structures. And because it is located so close to the site where those free radicals are produced, the mtDNA is especially vulnerable to being damaged by these free radicals. In the worst-case scenario, a free radical “hit” to the mtDNA can inflict damage that results in huge stretches of mtDNA being permanently deleted in one swoop when the mitochondrion next makes a new copy of its DNA. And because the stretch of mtDNA which is most prone to deletion mutations during aging encodes machinery that is essential to protein synthesis from any mitochondrial genes, the most common deletion seen in the mtDNA of aging cells can render a mitochondrion unable to make a single one of the 13 mitochondrially-encoded proteins that are critical components of their energy-generating system.
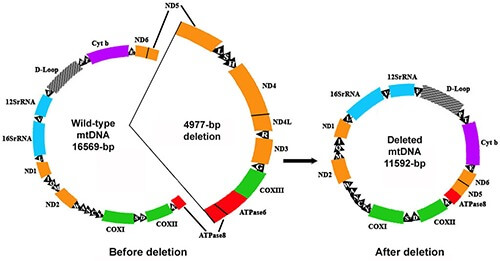
The 4977 base pair “common deletion” that accumulates in some aging cells encodes all of the mitochondrial tRNAs and the genes for seven subunits of the mitochondrial electron transport chain. Credit: Oncol Rev 13(1): 409.
Our cells have a system in place (mitophagy) for removing defective mitochondria, following which the remaining mitochondria are replicated to make up for lost numbers, so you wouldn’t think this would be a problem. But the mechanism that the mitophagy machinery uses to determine whether a given mitochondrion is damaged or not is unable to pick up on large deletions, giving them a kind of “cloaking device.” This has the perverse result that the cell tends to hang onto these defective, mutant mitochondria, while sending others with much more common but milder defects to the cellular recycling center. Mitochondria with deletion mutations thus have a selective advantage over healthy or mildly damaged mitochondria, with the result that once even one mitochondrion suffers a deletion, its progeny quickly take over the entire cell.
Only a few cell types in our body are vulnerable to this mutant takeover phenomenon, and only a small percentage of cells of those types actually suffer it during current lifetimes — but the harm those few cells inflict on our health and function belies their small numbers, for two reasons. First, the cell types that can be afflicted by such “clonal expansion” are critical cells that we can’t afford to go offline as we age because they don’t get replaced during our lifetimes, such as skeletal and heart muscle cells and brain neurons. Indeed, cells with mtDNA deletions feature prominently in some of the toughest diseases and disabilities of aging, such as Parkinson’s disease and sarcopenia (the aging disease that causes even master athletes to not only lose muscle mass as they age, but to suffer a loss of strength that is disproportionate to the amount of muscle lost).
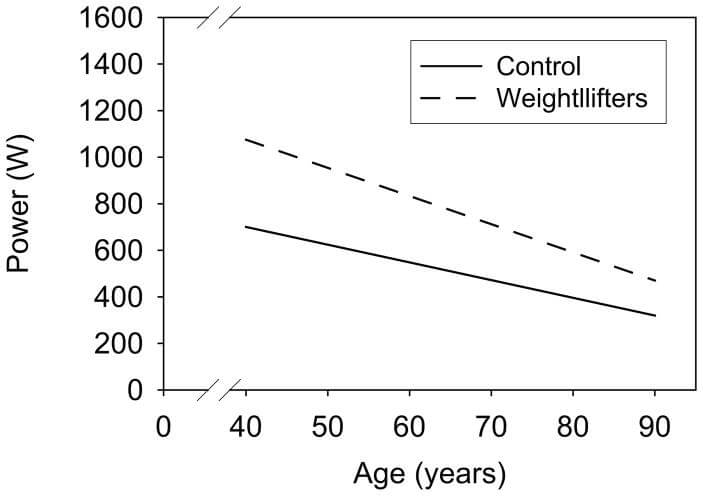
Both sedentary adults (solid line) and elite Master-class weight-lifters (dashed line) lose muscle power with age. Lifelong exercise sustains a higher level of strength and power, but the rate of loss may be faster in trained persons. Credit: Clin J Sport Med 18(6): 501–507, with data from Pearson et al., 2002.
Moreover, those few cells may wind up wreaking havoc not only in the tissues of which they are working units, but across the body as a whole. Because none of the mitochondria in such cells are able to produce cellular energy in the most efficient way, these mutant mitochondria enter into an abnormal metabolic state to keep going — a state that produces little energy, while generating large amounts of waste that the affected cell is not equipped to metabolize. Instead, such cells are thought to dump their mutant mitochondria’s waste into the circulation, causing oxidative stress to rise all over the body and deranging cellular signaling.
Rejuvenation Biotechnology Solutions
It would be ideal if we could prevent mitochondrial deletions from happening in the first place, or fix them directly after they’ve occurred; unfortunately, the state of biotechnology is nowhere near the point where either of these goals could be achieved in time to save people alive today. Instead, strategies for MitoSENS must start from the premise that some aging cells will inevitably be overtaken by large mitochondrial deletions (and many more will be riddled with less severe mutations of different kinds), and develop ways to engineer our cells with machinery that makes those mutations harmless to them and their neighbors.
Allotopic Expression: “Backup Copies”
The original SENS manifesto by SRF Founding CSO Dr. Aubrey de Grey and colleagues proposed one such strategy for obviating age-related mitochondrial mutations: inserting “backup copies” of the mitochondrial genes into the nucleus. Such “backup copies” would have to be engineered to be readable in the nuclear DNA code (which diverges from the code used by the mitochondrial original) so that they could be expressed from the nucleus and turned into proteins by the machinery in the cytosol — an outcome termed “allotopic expression” (AE). Such AE genes would also need additional sequences, borrowed from sequences already used by the existing nuclear-encoded mitochondrial genes, that would target them to the mitochondria. Some of them might also need to be tweaked in additional ways to make them easier for the mitochondria to import or to orient themselves properly in the mitochondrial inner membrane.
With such “backup copies” in place, the nuclear genome would be able to supply deletion-bearing mitochondria with the subunits of the ETS that are normally encoded in the mtDNA. With the missing components of their Complexes replaced through AE, the ETS in such mitochondria could still assemble and function just as it does when the same components are produced from the mitochondrially-encoded originals. With a complete and functioning ETS, our cellular power plants would continue humming along normally despite the loss of mitochondrially-encoded genes, and deletion-bearing mitochondria would lose the selective advantage that otherwise allows them to take over the cell and to drag it into the toxic, mutant metabolic state that threatens its neighbors. Additionally, in the safe harbor of the nucleus, such “backup copies” would be far less susceptible to mitochondrial or other free radical damage, and would be maintained by the more robust DNA repair machinery present in the nucleus.
In this case, we’re lucky: evolution has actually done the hardest part of this for us already, and given suggestions on how to finish the job. Today, the mitochondrial DNA contains instructions for building just 13 proteins needed by the mitochondria. But deep back in evolutionary history, there were literally about a thousand such genes. Those genes still exist, and they are all still needed to keep the mitochondria running normally — but over millions of years of evolution, natural selection drove the relocation of nearly all mitochondrial genes to the safer harbor of the nucleus. The nuclear-encoded mitochondrial proteins are constructed in the main body of the cell, outside of the mitochondria, and then imported into the mitochondria through specialized transport docks in their membranes.
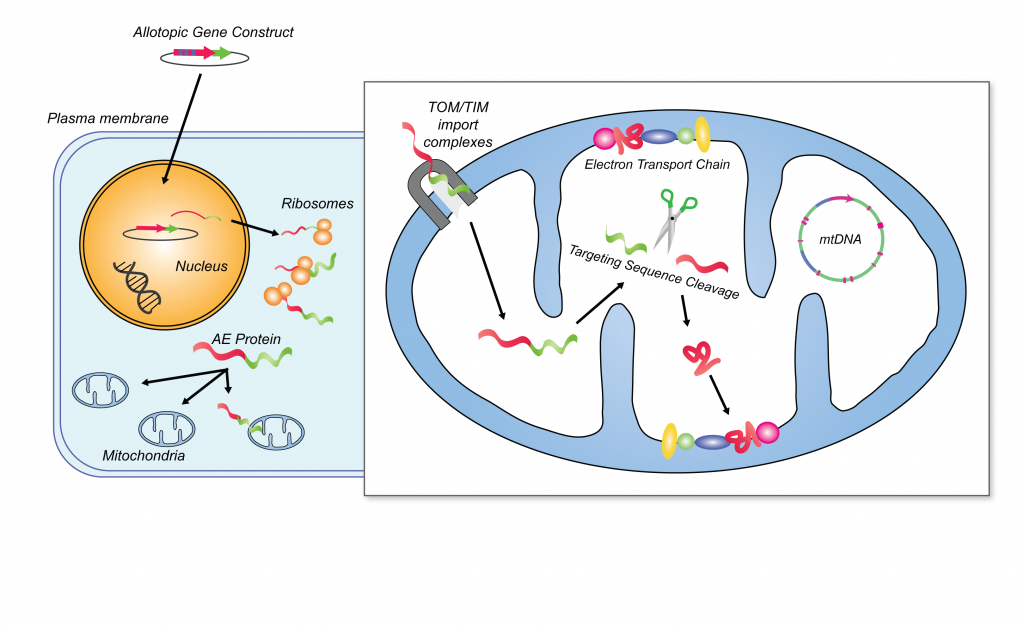
Allotopic expression. Credit: Human Gene Ther 19(12):1335-48, modified by Anne Corwin.
One of the challenges to allotopic expression is that the 13 remaining mitochondrially-encoded proteins are more challenging for the mitochondria to import than are the proteins whose genes have already migrated to the nucleus. This is because the electrical charges on the remaining 13 are such that they are repelled by the water in the cytosol; as a result, once they are formed in the main body of the cell, they quickly fold up on themselves, making a snarling skein that is difficult to thread through the pores in the mitochondrial membrane. This is probably why evolutionary pressure hasn’t already forced the genes to relocate.
But there are several potential ways around this problem. One is to look to solutions discovered by natural selection in other organisms. In several cases, one or a small number of unusual organisms have managed to transfer genes that are still encoded by the mitochondria in most animals into their nuclei — often by altering the charge of different regions of the protein, and less often by shortening or straightening the structure. It’s possible that suitably-modified versions of these organisms’ evolved solutions could be turned into an engineered solution for this part of the 13-part AE challenge.
Alternatively, we may be able to insert disposable molecular “braces” called inteins into the sequence of the proteins that would temporarily hold them straight enough to let them pass through the membranes. Following this, the imported protein would have to be further processed inside the mitochondria to excise the inteins and join the two functional halves together — a challenge, to sure, but well within the normal functional capacities of human cells.
A third approach, pioneered by Professor Marisol Corral-Debrinski at the Institut de la Vision at Pierre and Marie Curie University, Paris, was to add targeting sequences to the proteins that ensure that they are “decoded” from their genetic instructions into proteins very close to the mitochondrial surface, instead of far away in the cell body. This approach allows the allotopically-expressed proteins to be threaded directly into the mitochondria before they have the chance to twist up too much.
And as we’ll discuss below, the MitoSENS team at SRF has come up with additional technical tweaks to the core allotopic expression playbook that have enabled some of the greatest advances in AE in decades.
Gene Drive: The Genesis Device
To succeed, any engineering solution for mitochondrial mutations in aging must somehow get around the selective advantage of deletion-bearing mitochondria: otherwise, any re-engineered mitochondria in a cell will simply be outcompeted by the mutants. As we’ve discussed, allotopic expression bypasses the problem by delivering the ETS proteins to the mutant mitochondria instead of the genes for those proteins, which normalizes mitochondrial (and thus cellular) function even in the face of persistent mtDNA deletions.
In the early 2000s, Drs. Rafal Smigrodzki and Shaharyar M. Khan at the University of Virginia came at the problem from a completely different angle. They proposed to deliver replacement genes directly to the mutant mitochondria — and to ensure that the new strain would replace the old, they would engineer the replacement genomes with an enzyme that destroys the cell’s original mitochondrial genomes, clearing the field to allow its new seed to spread and thrive.
The term “gene drive” was brand new to biotechnology at the time, and Smigrodzki and Khan never used the term, but what they were proposing was a kind of gene drive for the mitochondria. To give the introduced mitochondrial genomes the ability to outcompete the cell’s pre-existing, damaged mtDNA, the therapeutic mitochondrial genome would include the gene for a kind of enzyme that would selectively destroy the existing mitochondrial genome but leave the engineered genome unscathed.
There are a few differences between the way that gene drives for whole organisms work and the way a gene drive for mitochondria would, which are related to the mitochondria themselves. These days, the main biotechnology used to fuel the dominance of an engineered genome in gene drives is the CRISPR-Cas9 system, but Smigrodzki and Khan’s idea predates the dawn of CRISPR, and involved the use of restriction enzymes instead.
Like the CRISPR-Cas9 system, restriction enzymes contain relatively long genetic segments that allow them to hone in on highly precise “recognition sequences” in the DNA of their target genomes, following which the restriction enzyme itself makes precise cuts in the offending DNA either at the recognition site or at a defined distance away from it. But for technical reasons, scientists have found it very difficult to work with CRISPR-Cas9 in mitochondria, so their original proposal makes more sense.
Additionally, many gene drives work by hijacking the cellular DNA repair machinery to replace the severed stretch of non-engineered, native DNA with a copy of the engineered gene sequence that is encoded on the opposite strand of the double helix of DNA. But mitochondrial DNA is single-stranded, so this specific approach won’t work. Instead, the mitochondrial gene drive relies on a “slash-and-burn” agriculture approach: clear the field by destroying the pre-existing mitochondrial genomes outright, and leave the engineered genome to spread across the genetic scorched earth.
As it happens, mitochondria contain several of the “restriction sites” that restriction enzymes target, so the first generation of mitochondrial gene drives would be engineered with restriction enzymes that target these naturally-occurring sites, which would allow them to destroy all copies of the cell’s original mtDNA, whether mutant or not. To prevent the restriction enzyme from attacking copies of the therapeutic replacement mitochondrial genome, each copy of the engineered genome would be altered so that it lacks the targeted restriction site.

A mitochondrial gene drive to overcome the selective advantage of deletion-bearing mitochondria. Credit: Anne Corwin.
Of course, at some point, the replacement genome would itself begin to suffer mtDNA mutations, and in some cases deletion mutations. So to allow the genetic Etch-a-Sketch to be periodically shaken clean again, successive generations of the mitochondrial gene drive would feature restriction enzymes that feature restriction enzymes that target sites that were not engineered out of the earlier generation’s genome — and the replacement genomes would in turn have new restriction sites soldered into them so that they, in turn, could be cleared out by yet another generation of the therapy when their time had come.
Thanks to these two core features (the restriction enzyme and the engineering out of the restriction enzymes’ targeted restriction site), the engineered mitochondrial genome is expected to act like the Genesis Device in Star Trek II: The Wrath of Khan, destroying the existing genomes in favor of the rapid takeover of what Spock called “its new matrix.”
Where We Are Now
The pace at which progress in this Strand of the SENS platform has accelerated in the short decade since SENS Research Foundation was founded can only be grasped by looking back at where we’ve been. When Ending Aging was published in 2007, only two groups had reported the allotopic expression of any mitochondrial genes in human cells — and one of those reports was doubted by many in the field.
The next major AE advances were made by Professor Corral-Debrinski, who developed a new trick to ensure that the AE proteins would actually get delivered to the mitochondria once expressed. As we discussed earlier, one of the main reasons why many of the 13 mitochondrial proteins we need to allotopically express have not yet been moved to the nucleus by evolutionary selection is that they are highly prone to curling up on themselves when expressed in the main body of the cell.In a kind of cellular onshoring decision, she modified the “zip code” attached to the working copies of her AE genes to direct them to production machinery close to the mitochondrial surface. This gave them a better chance of being imported before they had a chance to get hopelessly twisted up with themselves. She used this new approach in a key study demonstrating allotopic expression in the eyes of rats, which was enabled by SENS Research Foundation funding when housed as part of the Methuselah Foundation.
But there was another problem. While a number of groups continued to report versions of more mtDNA genes being expressed allotopically, nearly none of these groups found that their AE proteins improved the function of cells lacking a normal mitochondrial copy of the same gene, leading several groups to question whether allotopic expression could ever be made to work.
Happily, two major advances in the last decade have put most of the skeptics’ concerns to rest — and SENS Research Foundation played a key role in both of them.
First, close to a decade ago Professor Corral-Debrinski’s AE gene therapy was licensed by the biotech company Gensight, which used it as a treatment for patients with an inherited mutation in this gene that causes them to go blind. Gensight advanced her AE gene construct into a successful initial human clinical trial, which showed that the construct substantially improved function in the eyes of people afflicted with Leber’s hereditary optic neuropathy (LHON), an inherited mutation in one of several mitochondrial subunit genes (in this case, ND4) that can cause pathology in several organs, most often resulting in vision loss.
The results of the first trial were ambiguous, because of the substantial improvement in the patients’ “control” eyes (which had not been given the construct), but studies in cynomolgus monkeys and other animals suggest that this may have been due to “leakage” of the construct from the treated eye over to the untreated one. That trial was followed up by the larger REFLECT Phase III clinical trial, which confirmed that their mitochondrial gene therapy delivered long-term sustained effects in improving visual acuity in patients. Gensight is currently in the process of seeking approval with the European Medicines Agency (the EU’s version of the FDA), pending some work to scale and certify pharmaceutical-quality mass manufacture of the construct.
Second, after a decade of slow and painstaking experimentation, the MitoSENS team at SRF’s Research Center has been making rapid progress in the last six years. Their first important breakthrough came in 2016, with the successful AE of the mitochondrial ATP8 and ATP6 genes in human cells harboring a mutation from a patient with a severe ATP8 mutation. Other investigators had previously reported AE of both of these genes individually, but this was the first time two genes had been simultaneously AE’ed in the same human cells, the first time it had been done in a mitochondriopathy patient cell line, and also the first time AE had been convincingly used to rescue function in such cells (the key failure in previous reports of AE in human cells).
In the process of tinkering with their constructs to get them to work, the MitoSENS team discovered an important trick to boost the amount of mitochondrial subunit protein produced by cells from their AE genes. Recall that the DNA code “spells” the instructions for building proteins by recombining the basic four-base code of A, T, C, and G into different three-base combinations (“codons”), with a given three-base sequence designating a particular amino acid (the building blocks of proteins). But this allows for 64 different possible combinations, whereas there are only 20 actual amino acids for which they need to code. This creates space for a lot of redundancy in the code: for instance, there are four different synonyms for the amino acid valine.
When putting together their AE constructs, previous researchers had almost always simply used the most common genetic “spellings” for a given amino acid. But different “spellings” can speed up or slow down the machinery that turns genetic instructions into the resulting protein, and also affects the way that machinery treats other encoded instructions around it.
Part of the SRF MitoSENS group’s success with ATP8 was that for the first time, they designed their AE constructs using algorithms that selected the “spelling” for a given amino acid in the sequence based on the full range of what is known about the conditions under which mitochondria choose one “spelling” over another, to truly optimize the sequence. This turned out to work much better than the typical “minimally-recoded” method, leading to more ATP8 protein being produced from the construct, more protein localized in the mitochondria, and successful rescue of energy production from the patient cells’ mitochondrial ETS.
So next, the team tested the potential of this “codon optimization” algorithm to improve the allotopic expression of the remaining 12 mitochondrial genes. A quick-and-dirty test revealed that the codon-optimized constructs were more likely to lead to their encoded protein being produced at all than were constructs using the minimally-recoded method. And even in cases where both kinds of constructs were able to produce protein, the codon-optimized constructs produced significantly more protein than did the minimally-recoded versions.
Drilling down with a stronger test on one specific AE gene, the MitoSENS team put a codon-optimized allotopic ND1 construct head-to-head with a minimally-recoded version in a cell line derived from a patient with an ND1 mutation. The minimally-recoded version either produced no functional ND1 protein or wasn’t able to deliver it to the mitochondria in these cells. But the codon-optimized version not only produced viable protein that was imported into the mitochondria and had its import sequence cleaved, but it partially restored energy production in the mutant cells. The effect was not as powerful as the codon-optimized ATP8 had been in its mutant cell line, but it still demonstrated the importance of the new technique for successful AE.
The team then followed up two different AE constructs for the ETS subunit ND4, showing yet again that a codon-optimized version is more effective at restoring function to cells carrying the mtDNA mutation responsible for about 70% of cases of LHON than is a minimally-recoded construct based the one used by Gensight.
The MitoSENS team is currently working to further optimize constructs for the remaining gene, both in terms of additional refinements to the coding of the gene and in choice of the molecular “zip code” used to direct them into the mitochondria.
Allotopic Expression in Vivo
All of this work in improving if not yet perfecting allotopic expression, plus SENS Research Foundation’s parallel investment into the Maximally Modifiable Mouse (MMM) and enabling technologies that have emerged elsewhere in biotechnology, opened up the potential for a step-function advance in the technology: the successful allotopic expression of the mitochondrial ATP8 subunit across multiple tissues in living mice.
The MMM project sought to create a new solution for the delivery of rejuvenation biotechnology gene therapies that would eventually be translatable to humans. To test allotopic expression in vivo, the MitoSENS team crossed MMM mice with the only available mouse model harboring a mutation in the ATP8 gene that doesn’t die during embryonic development, namely the “mtFVB conplastic” mouse. These mice have a normal nuclear genome, but a minor mutation in their inherited ATP8 gene that leads to biochemical abnormalities in their cells and (in some reports but not others) minor problems in whole-body metabolic function.
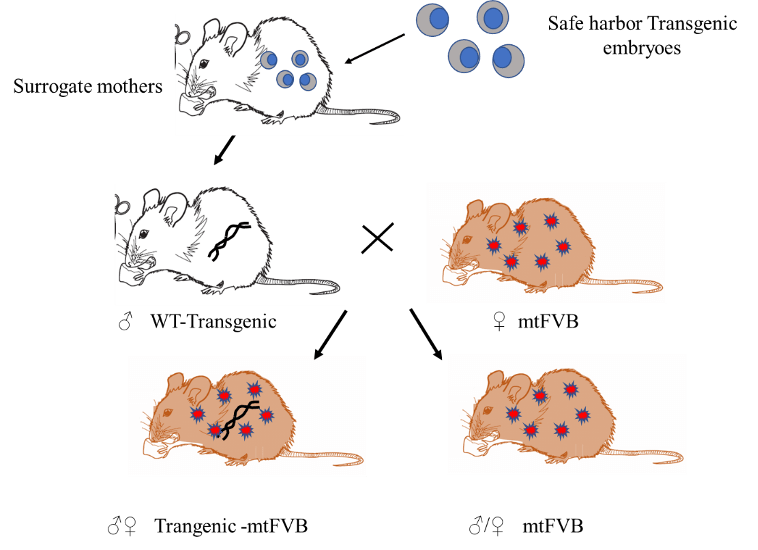
Using the Maximally Modified Mouse to test SENS Research Foundation’s ATP8 construct in the “mtFVB conplastic” mouse.
By using the MMM system to deliver the allotopic ATP8 construct into one group of mice with this mitochondrial defect and comparing them to another group of such mice with the same defect that did not receive it, the MitoSENS team could test whether their allotopic ATP8 would not only be expressed in the mice’s cells and delivered to the mitochondria, but whether it would successfully compete with the inherited mutant ATP8 protein to help restore normal biochemistry.
Excitingly, this use of allotopic expression in the actual tissues of living mice has so far passed every test. The was properly integrated into a “safe” place in the nuclear DNA, and the protein is expressed in multiple tissues. Once expressed, the allotopic ATP8 protein enters the mitochondria and slots into the cell’s energy-generating machinery. This is a critical step in the process of turning AE into a working rejuvenation biotechnology for humans.
SENS Research Foundation Research
As we have seen, SRF has both funded important work that enabled the development of what will likely be the first licensed mitochondrial gene therapy in humans, and has substantially revived the allotopic expression field with critical in-house work that has convincingly shown that it can work in cells. And now, our scientists appear to be getting AE to work in multiple tissues in living mice.
Our scientists are also making early progress on the gene drive strategy, as well as on an entirely different strategy that is in too early a stage to disclose, but that involves a potential way to tear the “cloaking device” off of mitochondria that bear large deletions. By exposing such cells to the mitophagy machinery, this approach could allow deletion-bearing mitochondria to be consigned to destruction instead of selectively surviving, and our cells would be able to rid themselves of these pernicious genomes. Depending on how successful it was, this approach could either greatly diminish our reliance on allotopic expression, or delay the need for it in order to initiate longevity escape velocity.
If successful, either of these alternative MitoSENS biotechnologies could offer us more tools in our toolkits to prevent cells from being overtaken by mutation-bearing mitochondria or to rescue them afterward. In addition to sheer optionality, these two strategies have the potential advantages of being faster to develop, easier to deliver, and with a lower bar for regulators to green-light clinical trials because they do not require gene therapy.